Pathologic Classification and Defining the Biological Potential
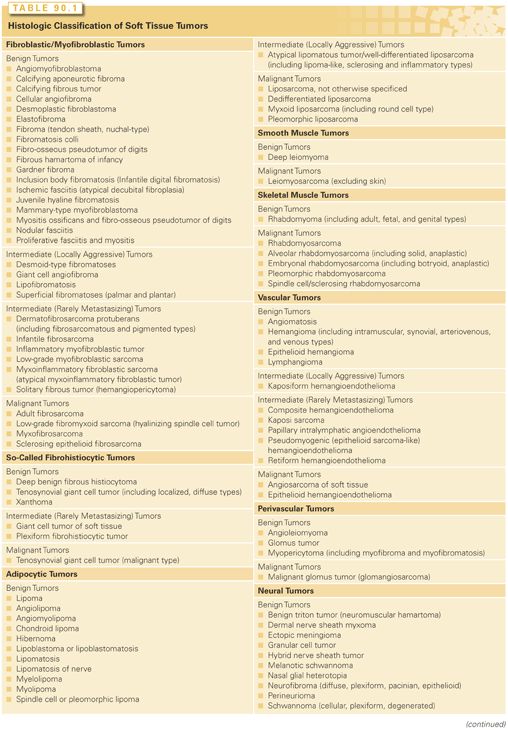
Soft tissue tumors, although clinically often nondistinctive, form a varied and complex group that may show a wide range of differentiation36 (Table 90.1). Their underlying histiogenesis has not been clearly defined. Except for subcutaneous lipomas or benign smooth muscle tumors, there is very little evidence that these lesions arise from their mature (differentiated) tissue counterparts. In fact, many liposarcomas arise at sites devoid of adipose tissue, and most rhabdomyosarcomas, which have molecular markers suggesting a myoid origin, develop in locations that lack voluntary muscle.
Soft tissue tumors may be benign, malignant, or borderline. The ratio of benign to malignant tumors is more than 100:1. Soft tissue tumors are notorious for the ease with which benign and malignant cases may be confused, particularly in small biopsy samples. Sarcoma histologic type is generally an important predictor of distinctive patterns of behavior and prognosis. Although many published series have combined all the histologic subtypes of sarcoma, the importance of such subtyping is exemplified by liposarcoma, in which the five histologic subtypes (well differentiated, dedifferentiated, myxoid, round cell, and pleomorphic) have totally different biologies and patterns of behavior.37–40 A further clear demonstration is the importance in pleomorphic sarcomas of myogenic differentiation, which is associated with a substantially increased risk of metastasis.41
As part of the World Health Organization classification of soft tissue tumors, it is now recommended to divide soft tissue tumors into four categories: benign, intermediate (locally aggressive), intermediate (rarely metastasizing), and malignant.36 Most benign tumors do not recur locally, and those that do recur usually are not locally invasive and can be cured with complete surgical excision. Intermediate, locally aggressive soft tissue tumors often recur locally and are associated with a locally infiltrative growth pattern. Lesions in this category, such as desmoids, do not have any potential to metastasize but typically require wide excision with a margin of normal tissue for good local control. Intermediate rarely metastasizing tumors are often locally aggressive and occasionally give rise to distant metastases. The risk of metastasis, usually to lymph nodes or lung, is typically <2%, but is not reliably predictable based on histology. Examples of intermediate rarely metastasizing tumors include plexiform fibrohistiocytic tumor and angiomatoid fibrous histiocytoma. Malignant tumors (soft tissue sarcomas), in addition to potential for local invasion and recurrence, have a significant risk of distant metastasis, ranging in most instances from 10% to 100%, depending on histologic type and grade. Some histologically low-grade sarcomas (myxofibrosarcoma, well-differentiated liposarcoma) have a metastatic risk of only 2% to 10%, but these tumor types may progress to more aggressive tumors, acquiring a higher risk of distant spread.
Clinical and Pathologic Features of Specific Soft Tissue Tumor Types
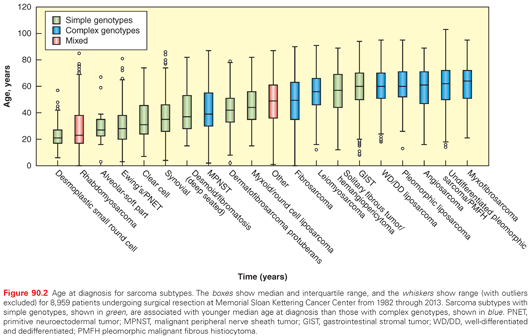
The distribution of adult soft tissue sarcomas by histologic subtype and anatomic site is shown in Fig. 90.1. Overall, the three most common histologic subtypes are liposarcoma, LMS, and UPS/PMFH.
Fibroblastic and Myofibroblastic Tumors
Fibroblastic and myofibroblastic tumors represent a very large subset of mesenchymal tumors. These lesions are generally composed of fibroblasts and myofibroblasts in varying proportions and may be confused with reactive or reparative processes or alternatively with malignant fibrosarcomas. In addition, some fibrous proliferations of infancy and childhood resemble lesions in the adult but have a better prognosis. With improved understanding of the molecular events that lead to formation of fibroblastic and myofibroblastic tumors, both low- and high-grade forms of the lesions have been reproducibly characterized, although a variety of names are still used to designate identical or overlapping entities.36 Among recent changes in the description of this subset of soft tissue lesions is the inclusion of dermatofibrosarcoma protuberans as a tumor of fibroblastic or myofibroblastic origin. The following sections summarize the features of the most common fibroblastic and myofibroblastic lesions with focus on sarcomas and those that may be mistaken for sarcoma.
Elastofibroma
Elastofibromas are rare, slow-growing benign tumors characteristically arising between the chest wall and the lower part of the scapula. They may occur bilaterally, and they may grow to large size. Rarely the tumors are observed at the infraolecranon area or near the ischial tuberosities. Elastofibromas have been considered reactive lesions, and they are thought to be associated with repetitive manual tasks. Up to a third of cases are familial, and bilateral subscapular lesions in this context can be diagnosed based on history and imaging alone. For unilateral spontaneous disease, biopsy is commonly employed; histologically, the lesions consist of swollen eosinophilic collagen and elastic fibers with associated fibroblast-like cells. The tumors have copy number alterations in both clonal and nonclonal patterns.42,43 If the diagnosis of elastofibroma is definitive, surgical resection can be reserved for the symptomatic patient.44,45
Fibroma
Fibroma is a nonspecific term usually applied to a group of poorly defined lesions in the skin or soft tissue. Management centers on complete surgical resection. While they can recur locally, metastases are not reported. Fibromas are characterized by a collagenous stroma with fibroblastic and myofibroblastic cells rarely demonstrating signs of atypia. While they can occur in unusual locations such as cardiac ventricle, they are more commonly observed in the skin, on the ovary, or associated with tendon sheaths. Nuchal-type fibromas, most commonly found in the posterior neck, are associated with male sex and diabetes mellitus; unlike common fibromas, nuchal-type fibromas can be infiltrative. Histologically similar are fibromas associated with Gardner syndrome, caused by germline APC mutation. These fibromas, which can be found on any area of the body, are commonly associated with metachronous desmoid tumors.
Nodular Fasciitis
Nodular fasciitis is a benign lesion usually seen in adults aged 20 to 40 years. The lesions typically grow rapidly over several weeks and reach 1 to 2 cm but rarely >5 cm. Pain and tenderness are common. The upper extremity is the most common site, especially the volar aspect of the forearm. Nodular fasciitis generally arises in the subcutaneous fascia or the superficial portions of the deep fascia. However, intra-articular fasciitis has been reported. Histologically, the lesions are nodular and nonencapsulated, showing plump mature fibroblasts arranged in short, irregular, or intersecting bundles; some lesions show hyalinization. Over 90% of these tumors have a genomic rearrangement affecting the USP6 gene, most commonly in the form of a MYH9–USP6 translocation.46,47 Because of their high cellular clarity, rapid growth, and high mitotic activity, these lesions are often confused with fibrosarcoma. They are, however, clinically benign; they generally regress spontaneously and recurrence after local excision is uncommon. Computed tomography (CT) and magnetic resonance imaging (MRI) characteristics are not pathognomonic, although they may show either a solid or some partially cystic changes, usually in the subcutaneous tissues.
Myositis Ossificans
Myositis ossificans is usually associated with trauma. The lesion is a benign disorder that histologically is composed of fibrous tissue and bone. Fibro-osseous pseudotumor of the digits may reflect a similar histopathologic entity. Despite its name, myositis ossificans is not necessarily confined to the muscle, nor is inflammation a prominent feature. The condition usually presents in athletic young adults as a tender soft tissue mass. Over a period of weeks, the mass usually becomes firm to hard. Radiographs show calcification several weeks after the lesion appears. Histologically the mass consists of fibroblastic tissue, often with prominent mitotic activity. Nonetheless, this process is benign and may be managed conservatively. It is important to distinguish between myositis ossificans and sarcoma, especially extraskeletal osteosarcoma. In general, myositis ossificans displays a more ordered growth pattern than osteosarcoma with cellular elements found in the center of the tumor and calcified regions almost exclusively in the periphery.
Superficial Fibromatosis
Superficial fibromatoses arise from the fascia or aponeuroses and generally are small and slow growing; they may be more common in patients with diabetes mellitus. Palmar fibromatosis is associated with flexion contractures (Dupuytren contracture) and is by far the most common form, affecting as many as one in five persons aged 65 years and older. This condition is more common in men than in women and tends to be familial. Although benign, these lesions tend to recur after simple excision. Plantar fibromatosis (Ledderhose disease) tends to occur in a somewhat younger age group but may occur with greater frequency in patients with palmar fibromatosis. Much less common is penile fibromatosis (Peyronie disease), which causes pain and curvature of the penis on erection. The fibrous mass in Peyronie disease primarily involves fascial structures, the corpus cavernosum, and rarely the corpus spongiosum. Peyronie disease is more common in men with palmar and plantar fibromatosis than in the general population. Any of the superficial fibromatoses may be managed by surgical excision, though more recently injection with collagenase or treatment with calcium channel blockers has been shown to have efficacy and may reduce the need for surgery.
Desmoid Tumor
Desmoid tumors (deep or aggressive fibromatosis) are rare lesions with an estimated incidence of two to four individuals per million. Histologically, desmoids are bland tumors, with uniform-appearing spindle cells in a densely hyalinized background. On a microscopic level, they are observed to infiltrate into surrounding normal tissues.48 Desmoids most commonly occur in the abdominal wall (particularly after pregnancy), in the mesentery of the small bowel, and in the extremity. Multifocal lesions are known to occur in a subset of young patients (often diagnosed between ages 10 to 25).26,49
Desmoids do not metastasize, but can be locally aggressive, causing pain, joint contracture, or bowel obstruction. Surgical resection has been the mainstay of therapy. However, compared to other soft tissue sarcomas, desmoids, especially those in the extremities or chest wall, have high local recurrence rates (~15% at 5 years). Nevertheless, individual desmoids exhibit a wide range of behaviors. While some tumors recur after multiple attempts at resection, some never recur, and it is now recognized that some desmoids remain stable in size without any type of intervention. In rare instances, desmoid tumors can cause death, which can result from local compression of vital structures (particularly in the head and neck), fistulization to the bowel by intraabdominal lesions, or injury related to aggressive attempts at resection.
Desmoids, in 85% of cases, have a mutation in exon 3 of the CTNNB1 gene that activates β-catenin.50 In a small proportion of desmoids, β-catenin is activated because of germline mutation in the APC gene; these desmoids occur in parallel with familial adenomatosis polyposis (classic or attenuated) in Gardner syndrome.51,52 Presence and site of CTNNB1 mutation has been reported to be associated with patient outcome,50 but this has not been validated and its use in prognostication is highly controversial.
Dermatofibrosarcoma Protuberans
Dermatofibrosarcoma protuberans (DFSP) is a generally indolent lesion characterized by translocation of the COL1A1 and PDGFRB genes.53 The resultant fusion protein activates platelet-derived growth factor (PDGF) receptor signaling, which likely underlies oncogenesis. The tumor, composed of mononuclear spindle cells, is a relatively monomorphous lesion. It involves both dermis and subcutis. DFSP is histologically similar to benign fibrous histiocytoma but grows in a more infiltrative pattern, spreading along connective tissue septa often with unpredictable radial extensions and multifocal nodules. The center of the tumor consists of uniform plump fibroblasts arranged in a distinct ordered pattern. Unlike fibrous histiocytoma, DFSP stains positive for CD34. Variants with melanin pigmentation (Bednar tumor), prominent myxoid stroma, myoid differentiation, and plaque-like morphology are recognized and may be confused with other types of soft tissue sarcomas.
DFSP is a rare sarcoma, but one of the most common cutaneous forms. This lesion typically presents in early or mid-adult life, beginning as a nodular cutaneous mass anywhere in the body. Growth is usually slow but persistent, and over many years it becomes protuberant and satellite nodules become visible. The gold standard of treatment remains surgery, though the infiltrative nature and multifocality of the lesion have historically led to high rates of local recurrence (up to 50% after simple excision in some series). However, when gross margins of ≥2 cm are planned, most tumors can be completely resected, and in the context of complete microscopic resection, 5-year local recurrence rates are ≤5%.54–56 Tumors with positive or close surgical margins in anatomically complex sites (e.g., near the brachial plexus) have an elevated risk of local recurrence, which is ameliorated with adjuvant radiotherapy.57 Postoperative radiotherapy is also indicated in the rare patient who has unresectable macroscopic disease. DFSP is sensitive to imatinib,58–62 probably because of constitutive activation of the PDGF receptor. The drug is now approved by the US Food and Drug Administration as the first line of treatment for advanced and metastatic disease. Metastasis (to lung or lymph nodes) occurs infrequently and generally only in the context of fibrosarcomatous degeneration, a high-grade form of the cancer.54,63
Inflammatory Myofibroblastic Tumor
Inflammatory myofibroblastic tumor (IMT) has gone under the names inflammatory pseudotumor and plasma cell granuloma, among others. Its histology is heterogeneous, with variable grouping of spindle, fibroblastic-myofibroblastic, and inflammatory-type cells. The variable appearance may result from the tumor’s genetic heterogeneity. While ALK rearrangements are common in the myofibroblastic component of the tumors,64,65 they occur in <70% of patients (almost exclusively in patients under 40). ALK is fused to a range of N-terminal partners, including RANBP2 and TPM3.66 Rarely, the HMGA2 gene on chromosome 12 is rearranged.67 Inflammatory components of the tumor do not have ALK fusions.68,69
IMT is a locally aggressive process. Many of the tumors occur in the thoracic and abdominal cavities, where they may appear as infiltrative or lobulated lesions on imaging studies.70,71 Size and location may cause symptoms related to compression of adjacent organs (e.g., bowel obstruction). Diagnosis of IMT is often difficult to confirm preoperatively; therefore, surgical resection may be both diagnostic and therapeutic. When complete resection is possible, it is curative in ~75% of cases. Metastasis is rare (<5%), but local recurrence is more common.72 Treatment of patients with advanced disease has been guided by the ALK fusions,73–75 and patients with ALK-positive advanced IMT have responded to crizotinib (according to Response Evaluation Criteria in Solid Tumors [RECIST]).76,77 In patients without an ALK fusion, treatment for locally aggressive disease is palliative, recognizing that few tumors have metastatic potential and the disease is rarely fatal (5-year survival of 87%). Of note, about 33% of patients with IMT develop a paraneoplastic syndrome including fever, growth failure, malaise, weight loss, anemia, and thrombocytosis.
Solitary Fibrous Tumor/Hemangiopericytoma
Solitary fibrous tumors (SFT) are identified by a prominent hemangiopericytoma-like branching vascular pattern. SFT encompasses fat-forming SFT and those lesions previously called hemangiopericytomas and giant cell angiofibromas, the latter containing giant multinucleated stromal cells and pseudo-vacuolar spaces. SFTs generally appear as slow-growing, well-circumscribed, painless masses. Histologically, they consist of tightly packed cells around thin-walled vascular channels of varying caliber. SFT cells stain for CD34 but not for factor VIII–related antigen. The lesions likely all have a NAB2-STAT6 fusion protein that results in aberrant activation of EGR1 target genes, including IGF2 and FGFR1.78,79
SFT may be found at any location in middle-aged adults (median age, 50). Occasional cases occur in children and adolescents. The adult form is most common in the thorax, pelvis, retroperitoneum, orbit, and lower extremity. Many SFTs are indolent and surgically cured, although some behave like other high-grade sarcomas. Risk factors for metastases include size >10 cm and mitotic rate >4 per 10 high-power fields.80 Rarely, patients present with a very large SFT and hypoglycemia (Doege-Potter syndrome), which is associated with production of a form of insulin-like growth factor (IGF) 2 by these tumors.81–84 SFTs are highly resistant to standard doxorubicin-based chemotherapy, but sunitinib and sorafenib have activity.85,86
Fibrosarcoma
Adult fibrosarcoma is uncommon (approximately 1% of adult sarcomas).87 The tumor typically has many copy number alterations, though these do not appear to affect a single driver oncogene or tumor suppressor.88 Fibrosarcoma is a malignant or intermediate (rarely metastasizing) tumor, composed of relatively homogenous spindle cells with variable collagen production and rare pleomorphism. Classical fibrosarcomas have a herringbone pattern on light microscopy. Like most soft tissue sarcomas with complex karyotypes, fibrosarcomas usually involve the deep tissues of the extremities, trunk, head, and neck of middle-aged and older adults. Some arise in the field of previous radiotherapy. Rarely, fibrosarcomas arise in the ovary or other unusual sites such as the trachea.
Infantile fibrosarcomas mimic the adult form histologically but are characterized by ETV6-NTRK3 fusion and appear to be more indolent than the adult-type tumor.89,90
Myxofibrosarcoma
Myxofibrosarcoma, formerly known as a myxoid variant of malignant fibrous histiocytoma (MFH), is a common type of soft tissue sarcoma. It usually occurs as a painless mass in the limbs and trunk of elderly adults. Histology shows a fibroblastic lesion with pleomorphism, a characteristic curvilinear vascular pattern, and at least a 10% myxoid component.41,91,92 Local recurrence occurs in up to 50% of cases, and is associated with tumors having a ≥75% myxoid component.93–96 However, a more recent study that treated 80% of patients with myxofibrosarcoma with adjuvant radiation found only a 14.6% local recurrence rate at 5 years and a 24% 5-year cumulative incidence of distant recurrence.97
Low-grade myxofibrosarcoma may progress to high grade in subsequent local recurrences and thus acquire a higher probability for metastatic spread. The most common sites of metastases are lung, bone, and lymph nodes. The overall 5-year survival is 60% to 70%.93–96 The 5-year disease-specific survival was 80% for patients presenting to Memorial Sloan Kettering Cancer Center (MSKCC) with primary myxofibrosarcoma of the extremity and trunk.
So-Called Fibrohistiocytic Tumors
The concept of fibrohistiocytic differentiation has been challenged, and it is now regarded as a poorly defined morphologic descriptor of histiocytic differentiation. The so-called fibrohistiocytic tumors, originally thought to arise from histiocytes that had fibroblastic potential, are almost certainly fibroblastic in origin. Thus, fibrohistiocytic is merely descriptive of their appearance; virtually none of these lesions show true histiocytic differentiation.98
Tenosynovial Giant Cell Tumor of Soft Tissue
The tenosynovial giant cell tumors of soft tissue encompasses entities previously called giant cell tumors of the tendon sheath, nodular tenosynovitis, or pigmented villonodular synovitis. These lesions contain multinucleate giant cells, siderophages, foam cells, and inflammatory cells. They are characterized by a t(1;2) translocation involving the colony stimulating factor 1 gene (CSF1) and most frequently COL6A3. This translocation leads to an increased level of CSF1, which is thought to modulate the inflammatory infiltrate noted in these tumors.99–101 Three forms of the lesions are recognized: nodular, diffuse, and malignant. Not surprisingly, the form is predictive of biological behavior.102 The nodular form, which has the greatest abundance of giant cells, is typically a well-circumscribed nodule in the digits. In contrast, the diffuse form has infiltrative borders and most commonly occurs in the large joints (e.g., knee, wrist, foot) or the surrounding soft tissue (e.g., thigh). They can be intra-articular. The malignant-type form demonstrates increased mitotic count and nuclear dysmorphism; these tumors likely have additional molecular alterations (other than COL6A3-CSF1 translocation) that drive disease progression. Malignant-type tumors may metastasize to lung or lymph nodes.
Surgery is the mainstay of treatment. The recurrence rate is just 10% to 20% for nodular tumors, but 18% to 50% for diffuse-type tumors. With adjuvant radiation, rates of local recurrence may be under 10%.103,104 Nevertheless, radiation is rarely used because of risk of joint fibrosis and secondary malignancies. Instead, recurrent or unresectable disease can be treated with a trial of imatinib, an inhibitor of the CSF1 receptor.105 Imatinib treatment results in disease stabilization in approximately 75% of patients. However, imatinib appears less effective in the rare diffuse-type tenosynovial giant cell tumors that have a malignant component.106 Because of their metastatic potential, malignant-type tumors should be managed aggressively as high-grade sarcomas. Multiple recurrent lesions that threaten limb integrity can be controlled with radiotherapy in both the tendon sheath and intra-articular forms.103
Fibrous Histiocytoma
Fibrous histiocytomas are benign tumors that usually present as solitary, slow-growing nodules, although up to one-third are multiple. Histologically, they consist of fibroblastic and histiocytic cells often arranged in a cartwheel or storiform pattern. When such lesions occur in the skin, they are often called dermatofibromas or sclerosing hemangiomas. Superficial lesions usually are cured by simple excision. Deeper lesions should be resected with a wider margin of normal tissue to prevent local recurrence.
In rare cases, fibrous histiocytomas are aggressive (“malignant dermatofibromas”). These lesions have a propensity for local recurrence, have been reported to metastasize, and, in a few patients, can cause death. Copy number alterations have been detected in these tumors, suggesting an underlying molecular aberrancy resulting in an invasive phenotype.107
Xanthoma
Xanthoma refers to a collection of lipid-laden histiocytes and is seen in diseases associated with hyperlipidemia. These lesions are generally cutaneous or subcutaneous but may involve deep soft tissues. Presumably, xanthomas are reactive lesions.
Adipocytic Tumors
Lipoma
Lipomas are the most common benign soft tissue neoplasm. They usually arise in subcutaneous tissue, most frequently in trunk and proximal limbs. Although deep-seated benign lipomas do occur in the mediastinum or retroperitoneum, most fatty neoplasms in the retroperitoneum should be approached surgically as atypical lipomatous tumor/well-differentiated liposarcoma. Most lipomas are soft, painless, slow-growing, and solitary; however, 2% to 3% of patients have multiple lesions that are occasionally seen in a familial pattern. Lipomatosis is a term applied to a poorly circumscribed overgrowth of mature adipose tissue that grows in an infiltrating pattern. Solitary lipomas are lobulated lesions composed of fat cells. They are well circumscribed, being demarcated from surrounding fat by a thin, fibrous capsule. Most subcutaneous, solitary lipomas show reproducible cytogenetic aberrations: translocations involving 12q13–15, rearrangements of 13q, or rearrangements involving 6p21–33.108 In spindle cell lipoma, mature fat is replaced by collagen-forming spindle cells; this lesion typically arises in the posterior neck and shoulder in men between the ages of 45 and 65 years. Spindle cell lipomas show consistent chromosomal aberrations of 13q and 16q.109 Local excision of lipoma and these variants is generally curative, with local recurrence after simple excision in no more than 1% to 2% of cases.
Intramuscular lipomas differ from their more superficial counterparts by usually being both poorly circumscribed and infiltrative (in ~90% of cases). Intramuscular lipomas typically present in midadult life as slow-growing, deep-seated masses most often located in the thigh or trunk. In a patient with a deep-seated fatty tumor, it is important to exclude atypical lipomatous tumor (see “Liposarcoma”), which tends to be more common than intramuscular lipoma.
Angiolipomas present as subcutaneous nodules, usually in young adults, and in >50% of cases are multiple. The most common site is the upper extremity. Angiolipomas rarely grow >2 cm, but they often are painful, especially during their initial growth period. Microscopically, these tumors consist of adipocytes with interspersed vascular structures. Myxoid and fibroblastic angiolipomas are recognized.
Hibernoma
Hibernoma is a rare, slow-growing, benign neoplasm that resembles the glandular brown fat of hibernating animals. The literature consists primarily of case reports. Most of these tumors arise within the thorax, though hibernomas of the trunk, retroperitoneum, and extremities are also reported. Excision is generally curative.
Angiomyolipoma
The term angiomyolipoma is used for a nonmetastasizing renal tumor composed of fat, smooth muscle, and blood vessels. Angiomyolipomas of the liver have also been described. Angiomyolipoma is more common in women than in men and is seen in patients with tuberous sclerosis, caused by germline mutations in TSC1 or TSC2. In addition, sporadic angiomyolipomas sometimes have mutation or loss of TSC1 or TSC2, which function upstream of mammalian target of rapamycin (mTOR) signaling.110–114 Thus angiomyolipomas may be sensitive to mTOR inhibitors.110,115–117 Although angiomyolipoma is usually well demarcated from normal kidney, it may extend into the surrounding retroperitoneum. Angiomyolipomas may be solitary or multicentric, and they may produce abdominal pain or hematuria. Wide excision is curative, but tumors that are asymptomatic and not enlarging may be observed.
Liposarcoma
Liposarcoma is primarily a tumor of adults, with a peak incidence between ages 50 and 65 years. It accounts for at least 20% of all soft tissue sarcoma in adults. Liposarcoma may occur anywhere in the body, although the most common sites are thigh and retroperitoneum. Liposarcoma can be divided into three main biological groups: (1) atypical lipomatous tumor/well-differentiated (ALT/WD) liposarcoma and dedifferentiated liposarcoma, (2) myxoid–round cell, and (3) pleomorphic. Each of these groups has distinctive morphology, natural history, and karyotypic and genetic aberrations, which can be of considerable help in diagnosis.
ALT/WD liposarcoma is a locally aggressive, nonmetastasizing, malignant mesenchymal neoplasm composed of proliferating mature adipocytes with significant variation in cell size and nuclear atypia, at least in foci. ALT/WD liposarcoma usually presents as a deep-seated, painless, enlarging mass that over many years can attain a very large size. ALT/WD liposarcomas can be divided into four main morphologic subtypes: adipocytic (lipomalike), sclerosing, inflammatory, and spindle cell. The characteristic cytogenetic abnormality detected in most ALT/WD liposarcomas is supernumerary ring and giant marker chromosomes with amplification of the 12q13–15 region. This region contains the known oncogenes CDK4, MDM2, and HMGA2, which were found amplified in 95%, 87%, and 76% of ALT/WD, respectively.118 Only 1 of 55 ALT/WD liposarcoma tumors had no 12q amplification. Location is an important predictor of outcome in patients with ALT/WD liposarcoma. Extremity tumors rarely recur and have essentially no mortality. In a series at MSKCC, all cases that recurred did so after 5 years and had a significant component of sclerosing morphology.119 In contrast, retroperitoneal and mediastinal tumors may recur repeatedly and eventually result in death from uncontrolled local effects; they may also dedifferentiate and metastasize. Dedifferentiation occurs in up to 20% of ALT/WD liposarcomas, with higher risk in deep locations such as the retroperitoneum. In a series of 99 patients with primary retroperitoneal ALT/WD liposarcoma, the 5-year disease-specific survival was 83% and 5-year probability of freedom from local recurrence was 54%.40
Dedifferentiated liposarcoma is defined as an ALT/WD liposarcoma, either primary tumor or recurrence, that shows abrupt transition to a region of nonlipogenic sarcoma at least several millimeters in diameter. Radiologic imaging typically shows coexistence of fatty and nonfatty solid components, which in the retroperitoneum may be discontiguous. Macroscopically, dedifferentiated liposarcoma consists of large multinodular yellow masses containing distinct nonlipomatous (dedifferentiated) areas, which are solid and often tan-gray. The dedifferentiated areas may contain areas of necrosis and hemorrhage. Dedifferentiated liposarcoma appears to have a lower risk of distant metastasis than other high-grade pleomorphic sarcomas. Nevertheless, among 65 patients with primary retroperitoneal dedifferentiated liposarcoma, 5-year disease-specific survival was only 20% and 3-year local and distant recurrence-free survival was 17% and 70%, respectively.40 Dedifferentiated liposarcoma, like ALT/WD, is characterized by ring or giant marker chromosomes and by amplification of the 12q13–15 region. Some other copy number alterations occur at substantially higher frequency in dedifferentiated liposarcoma than in ALT/WD and are thus associated with progression. These include losses centered at 3p14–21, 3q29, 9p22–24, 10p15, 11q23–24, 17q21, and 19q13, and gains at 17p11 and 20q11. Of these progression-associated copy number alterations, the most common, 11q23–24, was noted in 42% of dedifferentiated liposarcoma compared with 4% of ALT/WD. The 11q23–24 loss was associated with genomic complexity and distinct morphology, whereas loss of 19q13 predicted poor prognosis.118
Myxoid or round cell liposarcoma accounts for approximately 40% of liposarcomas. The tumor consists of small, evenly dispersed oval or plump cells with little cytoplasm in a myxoid matrix containing a variable number of fat cells. A small number of signet-ring cells and multivacuolated lipoblasts are often present but are not required for diagnosis. The myxoid–round cell subtype usually occurs in the deep soft tissues of the extremities; in >66% of cases it occurs in the thigh musculature.91 Rarely, myxoid–round cell liposarcoma may arise in the retroperitoneum or in the subcutaneous tissue. More than 90% of myxoid–round cell liposarcomas have a t(12;16)(q13-14;p11) translocation.120 High histologic grade, defined as ≥5% round cell component, is a predictor of worse outcome in localized myxoid–round cell liposarcoma. Patients with these high-grade lesions have a 5-year survival of 50%.37,121 In general, pure myxoid lesions (0% to 5% round cell areas) are considered low grade and are associated with a 90% 5-year survival. In contrast to other liposarcoma types, myxoid–round cell liposarcomas tend to metastasize to unusual sites in soft tissue or bone, with multifocal synchronous or metachronous spread to fat pad areas in the retroperitoneum and axilla occurring even in the absence of pulmonary metastasis.39,121,122 These lesions are also unusual among soft tissue sarcomas in their extraordinarily high response rate to radiotherapy123 and their substantial sensitivity to ifosfamide38 and to the DNA minor groove–binding drug trabectedin.124–126
Pleomorphic liposarcoma, as the name implies, is a pleomorphic, high-grade, highly malignant sarcoma containing variable numbers of pleomorphic lipoblasts. Mitotic activity is high, and hemorrhage or necrosis is common. Pleomorphic liposarcoma accounts for <5% of all liposarcomas. Most arise in patients older than 50 years and occur in deep soft tissue of the extremities (lower more frequently than upper). Clinically, they metastasize early to lung in >50% of patients, and these patients usually die within a short time. Pleomorphic liposarcomas typically have high chromosome counts, complex structural rearrangements, and multiple regions of significant copy number amplification and deletion.127 They appear to be somewhat sensitive to gemcitabine-based128 and ifosfamide-based chemotherapy.38
Smooth Muscle Tumors
Leiomyoma
Leiomyomas are benign smooth muscle tumors that are quite common in the uterus and the gastrointestinal (GI) tract. Leiomyoma may also occur deep within the extremities, abdominal cavity, or retroperitoneum. Their histologic appearance is benign, with uniform, spindle-shaped nuclei in cells that appear similar to those of normal smooth muscle. Immunohistochemically, the cells are positive for smooth muscle markers such as smooth muscle actin. Angiomyoma is a histologic subtype of leiomyoma that tends to develop on the extremity at ages 30 to 60. Leiomyomas in women often express estrogen and progesterone receptors, which appear to activate Wnt signaling to promote proliferation of tumor stem cells.129 These hormone-regulated tumors may regress at menopause. When they are symptomatic, hormone-regulated tumors can generally be cured by surgical resection; for hormone-receptor–negative tumors, surgery is the treatment of choice.
In three rare clinical scenarios involving symptomatic leiomyomas, management may be difficult. First, cutaneous leiomyoma is a form that arises from the piloerector muscles of the skin. The nodules most often arise on the extensor surfaces of the extremities, and they may follow a dermatomal distribution. Multiple painful tumors may be observed. Although these cutaneous leiomyomas are histologically benign, they frequently recur after surgical excision, and they are often so numerous that excision is not possible. Second, intravenous leiomyomatosis is a rare condition in which nodules of benign smooth muscle tissue grow within the veins of the myometrium and may extend into the uterine and hypogastric veins. Rarely, these tumors extend up the inferior vena cava into the heart. The third management challenge is with diffuse peritoneal leiomyomatosis, which often occurs in association with pregnancy. Compression of adjacent organs may cause obstruction, as in other instances of sarcomatosis.
Leiomyosarcoma
LMSs are malignant lesions that develop from the smooth muscle of blood vessels, visceral structures, or the uterine corpus. Soft tissue LMS usually occurs in middle-aged or older adults and is the predominant sarcoma arising from larger blood vessels. LMSs may arise in any location, but more than half are located in retroperitoneal/intra-abdominal and pelvic sites, most commonly the uterus (see Chapter 73). The tumors are composed of spindle-shaped cells with markers of smooth muscle differentiation such as SMA or desmin. Unlike leiomyoma, however, LMS has many mitotic spindles and may have nuclear and cellular pleomorphism. The LMS genome is characterized by instability and numerous copy number alterations. While most of the copy number alterations are not recurrent, loss of TP53 and PTEN tumors suppressors is common. Recent reports also demonstrate that recurrent mutations in the MED12 oncogene occur in LMS (at least in uterine lesions).130,131
LMSs can arise in any vessel and may present insidiously with signs of venous outflow obstruction or pain related to encasement of nearby nerves. LMS of the inferior vena cava can present with the Budd-Chiari syndrome.132 Treatment of choice is surgical resection. Arterial bypass may be performed. Venous reconstruction, however, is rarely successful, as vein grafts rarely remain patent for prolonged periods. Moreover, patients generally develop collateral veins during the months after resection. Therefore, venous reconstruction can be deferred even in the context of inferior vena cava resection.132 Tumor grade and size predict risk of disease-related death; retroperitoneal lesions are generally large and high grade, resulting in recurrence risks of >50%.133 Small LMS-like lesions in the dermis carry no discernable risk of metastasis, and the old term of “cutaneous LMS” has generally been replaced by “atypical intradermal smooth muscle neoplasms.”134
Skeletal Muscle Tumors
Rhabdomyoma
Nonmalignant tumors of striated muscle—rhabdomyomas—are rare but are clinically benign. They are subdivided into adult, fetal, and genital type lesions; cardiac rhabdomyomas are associated with the tuberous sclerosis syndrome. Adult and fetal rhabdomyomas are most commonly identified in the head and neck. Rhabdomyoma can be distinguished from rhabdomyosarcoma by its lack of nuclear atypia. Pathogenesis appears to be related to activation of the hedgehog pathway; fetal rhabdomyoma may occur in individuals with a germline mutation in the hedgehog inhibitor PTCH1, which is associated with basal cell nevus syndrome. Rhabdomyomas, if symptomatic, are managed with surgical resection.
Rhabdomyosarcomas
Rhabdomyosarcomas (malignant tumors showing skeletal differentiation) are aggressive malignancies that are the most common soft tissue sarcomas of infants and children. Treatment of rhabdomyosarcoma nearly always requires multimodal therapy, typically employing surgery, radiation, and chemotherapy based on a vincristine-dactinomycin-cyclophosphamide backbone.135,136 The recognized types are embryonal, alveolar, and pleomorphic.
Embryonal Rhabdomyosarcoma. Embryonal rhabdomyosarcoma is a small-cell tumor showing features of embryonic skeletal muscle. It usually arises in the orbit or genitourinary tract in children, though rare cases arise in adolescents and adults. The botryoid type, which frequently originates in mucosa-lined visceral organs such as the vagina and the urinary bladder, generally grows as a polypoid tumor. Genomic analysis demonstrates that these tumors have various copy number alterations that may directly or indirectly affect activity of oncogenes and tumor suppressors such as RB1, p53, RAS, hedgehog, FGFR4, Akt, ALK, Notch, and β-catenin.137–140 These findings have not yet led to clinically useful targeted therapies, but chemotherapy and radiation are very effective for pediatric embryonal rhabdomyosarcomas, even metastatic cases. Embryonal rhabdomyosarcomas in adults usually regress in response to pediatric chemotherapy regimens, but survival is worse for adults than for children.141 Factors strongly associated with poor prognosis in adults are metastatic disease at presentation and poor response to chemotherapy.142
Alveolar Rhabdomyosarcoma. Unlike the embryonal type, alveolar rhabdomyosarcoma is observed more commonly in adolescents and adults than in younger children. For younger children, alveolar rhabdomyosarcoma appears to have a worse prognosis than embryonal rhabdomyosarcoma. Histologically, the lesion is composed of ill-defined aggregates of poorly differentiated round or oval cells that frequently show central loss of cellular cohesion and formation of irregular “alveolar” spaces. They cytologically resemble lymphoma cells and show partial skeletal differentiation. In most cases, specific translocations create a PAX3-FOXO1 fusion gene (in the majority of patients) or a PAX7-FOXO1 fusion (in a smaller subset); rarely fusion-negative lesions are diagnosed.143,144 These fusion genes have prognostic significance (see Chapter 89), so fusion gene status, irrespective of histology, is a critical factor in risk stratification.145,146 Some tumors have amplification of the PAX7-FOXO1 fusion, which is associated with improved outcome though the underlying molecular mechanisms have not been delineated.147 As in embryonal rhabdomyosarcoma, gene amplifications result in activation of a range of oncogenes. These include FGFR4, ALK, CDK4, and MYCN.148
Pleomorphic Rhabdomyosarcoma. Pleomorphic rhabdomyosarcoma is the most common form of rhabdomyosarcoma in adults and can be associated with prior radiation. Histologically, the tumors have pleomorphic round cells and spindle cells with atypical nuclei and markers of skeletal muscle differentiation. They are characterized by complex copy number alterations. The prognosis for these pleomorphic tumors is poor, and in one series, 28 of 38 patients (74%) died of the disease.149 Treatment is surgical, but, because of the poor prognosis, eligible patients are given adjuvant radiation and chemotherapy. These tumors are less sensitive to systemic therapies than are embryonal or alveolar rhabdomyosarcomas, but some respond to anthracyclines and ifosfamide; in addition, anecdotes indicate some sensitivity to gemcitabine-based chemotherapy.128
Vascular Tumors
Hemangioma
Hemangiomas are among the most common soft tissue tumors. Many are present at birth and regress spontaneously; others are noted incidentally on imaging studies. They tend to be asymptomatic; however, some grow rapidly, impinge on vital structures, or cause consumptive thrombocytopenias. Imaging studies generally demonstrate a peripherally enhancing mass with lipomatous regions and spiculated calcium deposits. Management by observation is generally safe. For symptomatic disease, surgical resection is generally curative; sclerotherapy has recently been studied. Diffuse hemangiomatosis in the bone or lungs may be treated effectively with systemic interferon.
Epithelioid Hemangioendothelioma
Epithelioid hemangioendothelioma is a low-grade vascular lesion without structured vessels, but with tumor cells arranged in nests and cords. Epithelioid hemangioendotheliomas occur in the bone and associated soft tissues. In malignant disease, multifocal lesions are often observed, generally within the same limb. Metastases may occur in lung, lymph nodes, and bone.150 A fusion between the N-terminus of WWTR1 and the C-terminus of CAMTA1 occurs almost uniformly in these lesions, but not other vascular tumors.151,152 Surgical resection is the treatment of choice; however, multifocal disease often recurs. Nevertheless, <20% of patients die of disease. A phase 2 trial has suggested that treatment with bevacizumab may slow progression or induce partial response in the majority of patients.153
Angiosarcoma
Angiosarcoma is a malignant tumor composed of malignant cells that morphologically resemble endothelium to various extents. Angiosarcoma is currently considered to include those tumors previously termed lymphangiosarcoma because of their similarities in histology and outcomes.36 Most angiosarcomas develop in the skin or superficial soft tissue; <25% are in deep soft tissue.154 The disease occurs most commonly in the context of lymphedema (Stewart Treves syndrome) or after prior radiation (particularly radiation for breast cancer).16 Lymphedema-associated angiosarcoma presents difficulties for surgical planning because poor wound healing may compromise the ability to perform limb-sparing procedures. Another challenge is that lymphedema-associated angiosarcoma has high rates of both local and distal recurrence.155 Multicentric angiosarcomas on the scalp and face of elderly men typically show unrelenting progression, which can cause severe ulceration and infection and eventually metastasis. Features reported to be associated with poor outcome include patient age, tumor depth, and size,154,156,157 but in general death from disease is common, with the median time to disease-specific death as short as 3 years in some patient subsets. Surgical resection is rarely curative; however, angiosarcomas are relatively sensitive, at least for brief periods of time, to anthracycline-based chemotherapy and taxanes.158,159 The discovery of KDR mutations and amplification of MYC and FLT4 has led to the investigation of antiangiogenic therapies in this disease.160,161
Perivascular Tumors
Glomus Tumor
Glomus tumors are rare soft tissue lesions that are almost always benign.162 The tumors appear to develop from smooth muscle cells associated with the glomus body, a modified arteriovenous anastomosis in the skin involved in thermal regulation. Unlike most soft tissue tumors, glomus tumors can cause considerable pain. They are most commonly found in the distal extremities (subungual region, hand, wrist, and foot) of young adults, though extradigital lesions are reported.163 The appropriate treatment is complete local excision. Most patients have sporadic, solitary tumors, for which the underlying genomic alterations are unclear. However, about 10% of patients have multiple lesions, many of them familial. Some of these patients have heterozygous germline mutations in the GLMN or NF1 genes.164,165
Neural Tumors
Neurofibroma
Solitary neurofibromas are small, slow-growing cutaneous or subcutaneous nodules that usually arise during the third decade of life. Neurofibromas may occur in unidentifiable cutaneous nerves or in larger trunks. Within an identifiable larger nerve, they expand into a fusiform mass and often extend into soft tissue; they are well defined and they may be nodular. Histologically, they show spindle-shaped cells in a myxoid stroma that contains collagen fibers. Multiple neurofibromas may be associated with NF1 (von Recklinghausen disease), a common genetic disorder caused by an autosomal dominant mutation at the 17q11.2 locus and affecting 1 in 3,000 live births. Clinical features of NF1 include café au lait spots, pigmented hamartomas of the iris, and neurofibromas of several types. Cutaneous neurofibromas arise in the skin in all patients with NF1, with sizes varying from millimeters to centimeters, and some may be painful. Plexiform neurofibromas are larger lesions that affect the large segments of a nerve, thickening and distorting the nerve with greater dysesthetic pain. The difficult distinction is neurofibroma versus MPNST, which may develop in patients with NF1. MPNST is usually distinguished based on rapid growth and increasing symptoms, and is confirmed by biopsy.
Benign Schwannoma
Benign schwannoma, also called neurilemmoma, occurs most commonly in people between the age of 20 and 50 years. Common sites include the head and neck, the flexor surfaces of the extremities, and the paravertebral area of the retroperitoneum. The lesion grows slowly, and if superficial is usually small at the time of diagnosis, but it can reach large size in the retroperitoneum without symptoms. The tumor is usually encapsulated and consists of two components: an ordered cellular region (Antoni A area) and a loose, myxoid component (Antoni B area). Fortunately, diagnosis can often be made by percutaneous core or needle biopsy in patients with lesions in the retroperitoneum, where morbidity of operation is to be avoided. The cellular variant is the lesion most often seen late in life as a painless vertebral mass.166 Complete resection is curative in most patients.
Granular Cell Tumor
Granular cell tumor is a rare tumor, probably of neural origin. It typically presents in adults as a small, poorly circumscribed subcutaneous mass, commonly seen in the oral cavity, and it is only rarely malignant. Granular cell tumors have been seen in all parts of the body, including the pancreas and bile duct. They can occur in multiple sites. Metastases have been reported in approximately 2% of cases, although most reports are single cases.
Malignant Peripheral Nerve Sheath Tumor
MPNSTs are highly aggressive soft tissue sarcomas that rarely occur sporadically in the general population. They may, however, occur with a lifetime incidence of 8% to 13% in patients with NF1, an autosomal dominant tumor predisposition syndrome caused by germline mutations in the NF1 gene.11 Most MPNSTs are associated with major nerves of the body wall and extremities and typically affect adults in the third to fifth decades of life. The lower extremity and the retroperitoneum are the most common sites, but MPNSTs can arise anywhere in the body. These tumors originate from the nerve sheath rather than from the nerve itself. There is also an MPNST with rhabdomyosarcomatous elements, termed a triton tumor, suggesting that the Schwann cell may be the source of a variety of heterologous elements in nerve sheath tumors.167
Tumor cells are usually elongated, with frequent mitoses, and are arranged in a hypocellular myxoid stroma; pronounced atypia and epithelioid features are also characteristic. The majority of MPNSTs are high grade and characteristically stain for the S-100 protein. Weak S-100 staining in an MPNST is associated with undifferentiated tumors and a five-fold higher risk of distant metastasis.168 Tumor size and p53 expression remain the most important independent predictors of disease-specific survival.168,169 Two recent studies have suggested that patients with NF1-associated MPNST have a worse outcome compared to patients with sporadic MPNST,170,171 and in one study this outcome difference was independent of tumor size.171
Complete surgical resection with or without adjuvant radiotherapy remains the most important treatment for those patients with primary disease. The role of neoadjuvant chemotherapy for patients with large primary and locally recurrent MPNSTs remains controversial. Overall response rates to chemotherapy are 21%, with improved outcomes noted when ifosfamide is added to adriamycin regimens.172 With an increasing understanding of the signaling pathways activated in MPNSTs, sorafenib, which targets the mitogen-activated protein kinase pathway, has been tested in single cases and in a histology-specific clinical trial.173,174 Patients with NF1 may be difficult to evaluate, independent of the features of the tumor itself. Staging and follow-up assessments are confounded by the detection of other nodules and masses that, although generally representing benign neurofibromas, need to be distinguished from recurrent local or metastatic disease or a second neurogenic sarcoma.
Extraskeletal Chondro-Osseous Tumors
Extraskeletal Osteosarcoma
Extraskeletal osteosarcomas are rare, high-grade sarcomas defined by production of malignant osteoid and bone. By definition, they are not attached to the skeleton. Unlike typical osteogenic sarcoma of bone, these tumors rarely occur before age 20, and most patients are older than 50 years. Most extraskeletal osteosarcomas arise in the extremities, although they have been reported in other sites, including breast, retroperitoneum, urinary bladder, and other visceral organs. Similar to those osteosarcomas arising from bone, extraskeletal osteosarcomas are highly heterogeneous on the microscopic level. Giant cells are a common feature, but no recurrent genomic events have been characterized. Surgical resection is generally used as single-modality treatment in this disease. Unlike osteogenic sarcoma arising from bone, extraskeletal osteosarcoma is not generally treated with adjuvant chemotherapy, although at least one series indicates a better outcome for patients treated with agents usually employed for classic osteogenic sarcomas.175
Tumors of Uncertain Differentiation
For most tumors in this category, there is no clear consensus as to the line of differentiation. However, for some tumors, such as synovial sarcoma and clear cell sarcoma, a line of differentiation can be clearly delineated, but no cellular counterpart in normal mesenchymal tissues can be defined.
Myxoma
Intramuscular myxoma is a rare tumor that occurs in adults, usually in the large muscles of the extremities. Myxomas consist of spindle cells without nuclear atypia. The stroma is composed of abundant myxoid tissue. The tumors are not highly vascular and do not enhance on MRI. Increased cellularity has been noted in a subset of lesions, termed “cellular myxomas.”176 Because of the high myxoid component and subset of cellular lesions, myxomas can appear similar to myxofibrosarcomas on biopsy. An aid to diagnosis is mutations in the GNAS1 gene, which are common in spontaneous myxoma as compared to myxofibrosarcoma.177,178 Multiple intramuscular myxomas occur in association with fibrous dysplasia (Mazabraud syndrome).
Angiomyxoma
Aggressive angiomyxoma is a soft tissue tumor generally identified in the pelvis or perineum of middle aged and older women. The tumors have a highly myxoid stroma with significant vasculature and small spindle or stellate cells without nuclear atypia. Aggressive angiomyxomas typically can slowly grow to large size and generally do not cause obstructive symptoms. Local recurrence is common after surgical resection and can result in considerable morbidity, given the location of these tumors, but distant metastases do not occur. Tumors express high levels of estrogen and progesterone receptors, and advanced disease may be managed with gonadotropin-releasing hormone agonists such as leuprolide. The tumor-initiating cell for angiomyxoma has not been characterized, but rearrangement of HMGA2 has been observed and appears to be associated with high expression of this oncogene.179
Neoplasms with Perivascular Epithelioid Cell Differentiation (PEComas)
The family of tumors known as PEComas are associated with neoplastic cells within the walls of blood vessels in the tumors. PEComas include clear cell “sugar” tumors of the lung, angiomyolipomas, and lymphangioleiomyomatosis (LAM). LAM is characterized by progressive interstitial infiltration of lungs by smooth muscle cells, resulting in cystic changes. It is a rare, progressive cystic lung disease predominantly affecting younger women of reproductive age. In end stages, LAM has been treated by lung transplantation.180 Angiomyolipomas are benign lesions that may grow quite large, but can safely be observed in most clinical scenarios (see “Adipocytic Tumors”). While angiomyolipomas or LAM do not metastasize, malignant forms of PEComa occur. PEComas such as angiomyolipomas or LAM are found in patients with tuberous sclerosis, and thus it is not surprising that TSC2 or TSC1 mutations or deletions are also found in sporadic lesions.110,113,181,182 Surgical resection may be sufficient to manage isolated tumors, but for metastatic disease or unresectable lesions, inhibitors of mTOR can be an effective treatment.115,183–185
Synovial Sarcoma
Synovial sarcoma is a spindle cell tumor with varying extents of epithelial differentiation, including gland formation, and is genetically distinct based on specific chromosomal translocations. Synovial sarcomas may be diagnosed at any age but the majority occur in young adults, between 15 and 35 years of age, and more commonly in males.186 Over 80% arise in deep soft tissue of the extremities, with about 50% in the lower limbs and most of the remainder in the upper limbs. Synovial sarcoma generally does not originate from synovial tissue, and it may be encountered in regions without apparent relationship to synovial structures, including the head and neck (<10%), thoracic and abdominal wall (<10%), or intrathoracic sites. Synovial sarcoma usually presents as a slow-growing mass with or without pain. Histologically, it may be monophasic or biphasic (i.e., composed of two morphologically distinct types of cells). Biphasic tumors have a characteristic pattern of epithelial cells surrounded by a spindle cell or fibrous component. Monophasic synovial sarcomas may be either fibrous or epithelial type, although the epithelial type is extremely rare. Calcification, with or without ossification, is seen in up to 10% of tumors, and synovial sarcoma may be confused with other calcifying tumors (e.g., thyroid neoplasms, which may exhibit calcification). The spindle cells stain positive for keratin, epithelial membrane antigen, and vimentin. S-100 staining may give positive results.
Nearly all synovial sarcomas contain a chromosomal translocation, t(X;18)(p11.2;q11.2).187 With the observation that 100% of biphasic and 96% of monophasic synovial sarcomas possess this translocation, it has become the gold standard in diagnosing synovial sarcoma.188 The translocation fuses the SS18 (SYT) gene with either SSX1, SSX2, or SSX4; the specific SSX gene involved may have prognostic significance.189,190 A recent study has suggested that metastasis in both pediatric and adult synovial sarcoma is strongly associated with genome complexity and with a gene expression signature related to mitotic control.191
Extraskeletal Myxoid Chondrosarcoma
Extraskeletal myxoid chondrosarcoma (EMC) is a malignant tumor characterized by a multinodular growth pattern and by chondroblastlike cells arranged in cords, clusters, or delicate networks within an abundant myxoid matrix. It occurs most commonly in the deep soft tissues of the proximal extremities and limb girdles in patients older than 35 years; two-thirds of patients are male. In contrast to the more common skeletal chondrosarcoma of bone, EMC seldom contains mature cartilage, and there is no convincing evidence of cartilaginous differentiation. Ultrastructurally, EMC is characterized by densely packed intracisternal microtubules and prominent mitochondria, whereas these are not apparent in skeletal chondrosarcoma. In addition, a nonrandom reciprocal translocation t(9;22), fusing the EWSR1 and NR4A3 genes, is present in about 50% of EMCs192–194 and is not seen in skeletal chondrosarcoma, which supports the idea that the two diseases have different molecular lineages. A second subgroup of EMC is characterized by a t(9,17) translocation joining TAF15 and NR4A3.195 EMCs usually grow slowly and long survival is typical, even in patients with metastases, which usually occur in the lung.196 Nevertheless, with prolonged follow-up, late local recurrence and metastasis are common. EMC is generally resistant to standard chemotherapy,197 but sunitinib has produced significant tumor regressions in two patients with advanced EMC.198
Alveolar Soft Part Sarcoma
Alveolar soft part sarcoma (ASPS) is a rare tumor comprising <1% of soft tissue sarcomas. The tumors are poorly circumscribed lesions that are composed of large epithelioid cells with abundant eosinophilic cytoplasm. The cells are generally arranged in a pseudoalveolar pattern with a highly vascular surrounding stroma. ASPS harbors a t(17-X) (p11.2;q25) translocation, resulting in the highly specific ASPSCR1-TFE3 fusion protein.199 Like most translocation-associated sarcomas, ASPS is most commonly diagnosed in young adults. Females outnumber males, especially among patients younger than age 20.200,201
ASPS often presents in the lower extremities, most commonly the thigh,201,202 as a painless mass. The tumor grows slowly, and patients may remain asymptomatic over years, even with metastatic disease.203 Local recurrence after surgery is rare, but ultimate prognosis is poor because ASPS characteristically metastasizes early and is essentially impervious to standard chemotherapy agents. In a large study from MSKCC, the survival rate of patients without metastases at diagnosis was 60% at 5 years, 38% at 10 years, and 15% at 20 years.201 The ASPSCR1-TFE1 fusion protein activates transcription of the MET oncogene, which drives growth of ASPS cells.204 Ongoing trials are examining the efficacy of MET inhibitors such as crizotinib in this tumor.205 The tumors also overexpress angiogenic receptor tyrosine kinases, and indeed targeted inhibitors such as sunitinib and bevacizumab have some efficacy for ASPS.206–208
Epithelioid Sarcoma
Epithelioid sarcoma, characterized by epithelioid and less commonly spindle-shaped cells, generally arises in the extremities. It occurs in two forms: distal-type (conventional) epithelioid sarcoma, occurring most commonly on the volar aspects of the hands and feet, and proximal-type, occurring most commonly on the perineum, groin, thigh, buttock, or less commonly the axilla. The proximal-type variant consists of large epithelioid carcinoma-like cells with pronounced cytologic atypia and prominent nucleoli frequently exhibiting rhabdoid features.209 It often grows in a multinodular pattern. Both proximal- and distal-type epithelioid sarcomas often have central regions of necrosis when examined histologically. Tumors located in deep tissue may spread along fascia planes, and thus epithelioid sarcoma requires extensive wide excision for complete tumor removal. Epithelioid sarcoma is also one of the few sarcomas in which lymph node metastases are fairly common, occurring in 20% of patients. Gross nodal disease should be biopsied, and if disease is present but the patient has no apparent distant metastases, a complete lymph node dissection should be considered. The role of sentinel node biopsy is highly debatable, with no proven effect on outcome.
Prognosis for patients with epithelioid sarcoma is generally poor. In a recent series that included 54 patients with localized disease,210 the 5-year local recurrence–free survival was 54%, distant recurrence–free survival 53%, and overall survival 62%. Independent predictors of worse survival were higher grade and deep location. Epithelioid sarcoma is moderately sensitive to chemotherapy, although responses are typically short lived. Compared with the distal variant, the proximal variant is associated with a more aggressive clinical course, resistance to radiation and chemotherapy, and worse disease-specific survival.209,211–214 Genetic analysis of epithelioid sarcoma is beginning to define molecular mechanisms for pathogenesis. Aberrations that have been seen include loss of the SMARCB1 tumor suppressor and upregulation of the epidermal growth factor receptor and mTOR pathways. Combined inhibition of these two pathways inhibited epithelioid sarcoma cell growth in vitro and in a xenograft model.215,216
Clear Cell Sarcoma (Melanoma of Soft Parts)
Clear cell sarcoma, initially described by Enzinger,217 is a sarcoma with melanocytic differentiation, typically involving the tendons and aponeuroses of young adults. The lesions are composed of epithelioid-type cells clustered in nests, each surrounded by collagenous bands. Clear cell sarcoma presents as a slowly growing soft tissue mass. Up to 50% of patients have pain or tenderness. Because its cells contains melanin and it tends to metastasize to regional nodes, clear cell sarcoma is considered to behave more like a melanoma than a soft tissue sarcoma. Genomic profiling and cluster analysis has also grouped these lesions more with melanoma than with sarcomas.218 However, unlike melanoma, clear cell sarcoma typically has a chromosomal translocation. In >75% of cases, this is t(12;22), fusing the EWSR1 and ATF1 genes.219 The fusion product activates the kinase MET, giving hope that MET inhibitors will have activity against this group of tumors.220
The treatment of choice is surgical resection. Gross disease in the lymph node basin is removed in tandem with wide resection of the primary tumor. Given the propensity of this subtype to nodal metastasis, sentinel node biopsy can be considered, though its clinical utility is debated.221 Size is a prognostic factor in outcome, and the majority of tumors are <5 cm at diagnosis. Metastasis is common, and 5-year survival approaches 50%. Chemotherapy has limited benefit, with platinum-containing regimens offering the most potential benefit, though recent reports suggest that antiangiogenic treatment (e.g., sorafenib and sunitinib) may have activity.222,223
Desmoplastic Small Round Cell Tumor
Desmoplastic small round cell tumor is composed of monotonous blue cells as stained on hematoxylin and eosin stain. The cells have little cytoplasm and may be arranged in nests or in an infiltrative pattern within a prominent desmoplastic stroma.224 The tumors are characterized by a specific t(11;22) translocation, fusing the EWSRI and WT1 genes.225,226 The disease usually arises in children and young adults, in whom abdominal sarcomatosis is a common presentation. For this reason, prognosis is generally poor and management can be difficult. In a review of 40 histologically proven cases, only 30% of patients were alive at 3 years from diagnosis.227 Surgical resection is possible for isolated tumors, but more commonly, patients are managed with front-line chemotherapy followed by debulking. Factors associated with improved overall survival are gross total resection and good responses to chemotherapy agents, such as that used for Ewing sarcoma.227,228
Undifferentiated/Unclassified Tumors
High-Grade Undifferentiated Pleomorphic Sarcoma/Pleomorphic Malignant Fibrous Histiocytoma
MFH was originally defined as a malignant pleomorphic spindle cell tumor with fibroblastic and histiocytic differentiation. However, pathologists now agree that this morphology may be shared by a wide range of malignancies.98 Careful immunohistochemical and histopathologic analysis showed that many sarcomas previously classified as pleomorphic MFH had a specific line of differentiation and could be reclassified as myxofibrosarcoma (30%), myogenic sarcoma (30%), myofibroblastic sarcoma (11%), liposarcoma (4%), soft tissue osteosarcoma (3%), or malignant peripheral nerve sheath tumor (2%), whereas only 16% had no specific line of differentiation.41 Thus, the term UPS is now reserved for pleomorphic sarcomas that by current technology show no definable line of differentiation.41,92,98 UPS characteristically is a tumor of later adult life with peak incidence at ages 60 to 70. UPS usually presents as a painless, deep-seated mass; the most common site is the lower extremity, followed by the upper extremity. A subset of UPSs arise at the site of prior radiotherapy24 and very rare cases arise at the site of chronic ulceration. About 5% of patients present with metastasis, typically to lung. Clinical and pathologic studies have shown a remarkable degree of heterogeneity of morphologic and biological features, prognosis, and treatment response. UPS typically has an aggressive clinical course, with many patients developing metastatic disease within 3 years of diagnosis. The 5-year disease-specific survival was 65% for patients presenting to MSKCC with primary UPS of the extremity and trunk.
Histologic Grading
After establishing the diagnosis of sarcoma, the most critical piece of information the pathologist can provide to the clinician is histologic grade. Grading, based on morphologic features only, evaluates the degree of malignancy and predicts outcomes, mainly the probability of distant relapse. The pathologic features that define grade include mitotic index, necrosis, cellularity, pleomorphism, and histologic type and subtype or differentiation; the two most important factors seem to be the mitotic index and the extent of necrosis.91,229 Unfortunately, the criteria for grading are neither specific nor standardized, and there is no general consensus on the morphologic criteria to use. Several grading systems are used: a four-grade system (Broders),230 three-grade systems such as the National Cancer Institute (NCI) grading system231 and that of the Sarcoma Group of the French Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC),232 and a two-grade system as is used at MSKCC.233 All of these grading systems have proven to correlate with overall survival and disease-free survival.
In general, the two most widely used grading systems are the NCI system231 and the FNCLCC system.234 Both are three-tier systems (high/intermediate/low). A comparative study showed that the prediction of distant metastasis and tumor mortality was slightly better with the FNCLCC system than with the NCI system.232 However, two studies have evaluated the interobserver reproducibility of the FNCLCC system and showed only 60% to 75% agreement on tumor grade and 61% to 75% agreement on histologic type.235,236 This high level of disagreement (25% to 40%) even among expert sarcoma pathologists emphasizes the importance of histologic peer review and the need for more objective systems for sarcoma grading and classification.235–237 In fact, neither the FNCLCC nor NCI system has been formally endorsed by either the World Health Organization91 or the Association of Directors of Anatomic and Surgical Pathology.238
In the MSKCC system, grade is classified as high or low based on the degree of mitotic activity, necrosis, cellularity, and tumor differentiation.233 Grade in this system has excellent correlation with clinical outcome in many histologic types and has proven to be one of the most important independent predictors of disease-specific survival.239 In addition, this system avoids the management dilemma of an “intermediate” grade, which in most institutions would be lumped with and treated as high-grade sarcoma, potentially resulting in overtreatment. The authors recognize that in certain situations (approximately 5% to 10% of cases), the distinction between low- and high-grade tumors can be quite difficult, and therefore an intermediate grade would seem the most suitable. These difficult cases can be graded most appropriately by using systematic sampling and thorough examination.
Although there is widespread use of some form of grading system in the diagnosis and management of sarcomas, there is also agreement that no current grading system performs well for every type of sarcoma. For multiple reasons, certain histologic types of sarcoma do not lend themselves well to grading. An example is myxoid/round cell liposarcoma, where round cell histology in just >5% of tumor area is sufficient to predict high-grade behavior, with a >50% risk of distant metastasis.121 In addition, unequivocal characterization of grade is difficult in large lesions, especially in tumors that can reach 2 or 3 kg.
The lack of standardization on grading has obvious clinical implications. In adjuvant chemotherapy trials, high grade is defined differently at different centers, which makes it hazardous to compare results between trials or to combine results of multiple trials. For example, tumors of 240 patients who participated in the Scandinavian Sarcoma Group adjuvant trial for high-grade extremity sarcoma were reviewed by a panel of reference pathologists. Eligibility was limited to patients with grade 3 or 4 sarcomas in a four-grade system. On review, 5% of the patients were considered ineligible because their tumors were low grade.235 In addition, the original pathologists and the reference pathologists had considerable discordance with regard to whether a lesion was grade 3 or 4. Although the adjuvant regimen did not affect survival, a difference in survival was noted between patients with tumors of these two grades as assigned by the reference pathologists.
Grading needs to be adapted to the modern management of patients with sarcoma, who often undergo a limited core biopsy rather than an open incisional biopsy. Grading on such limited material needs to be complemented with imaging and molecular data. Extent of necrosis may best be evaluated by imaging studies because they enable macroscopic examination of the entire tumor. Both MRI and magnetic resonance spectroscopy have been used to assess necrosis, chemotherapy response, and grade in sarcoma. Mitotic index is difficult to determine on limited core biopsy material, and MIB-1 (Ki-67) scores of proliferation may be more reproducible240 and have better predictive value than grading using mitotic index.241 Other molecular characteristics, such as mutation or nuclear overexpression of TP53 and high Ki-67 proliferation index, are associated with high grade and poor survival.242 A gene expression signature of 67 genes related to mitosis and chromosome management, the Complexity Index in Sarcomas was an independent predictor of metastasis outcome even adjusting for histologic subtype and FNCLCC grade and is superior to the FNCLCC grading system in determining metastatic outcome for sarcoma patients.243,244
Clinical Features
The presence of soft tissue sarcoma almost invariably is suggested by the development of a mass. This mass is usually large, is often painless, and may be associated by the patient with an episode of injury. Approximately one-third present with a size <5 cm, one-third with a size 5 to 10 cm, and one-third with a size >10 cm. The focus of the clinical evaluation is to determine the likelihood of a benign or malignant soft tissue tumor, the involvement of muscular or neurovascular structures, and the ease with which biopsy or subsequent excision can be performed. Size becomes an important feature (see “Prognostic Factors”), and definitive diagnosis depends on biopsy results and histologic confirmation.
Differential Diagnosis
The differential diagnosis of a soft tissue mass includes, in addition to sarcoma, a variety of benign lesions, as well as primary or metastatic carcinoma, melanoma, and lymphoma. The major concern when confronted with a soft tissue mass is determining whether the lesion is benign or malignant. In most patients with small lesions, or even on occasion large lesions, the important distinction is lipoma, the most common soft tissue tumor, versus other tumors. Most benign lesions are located in superficial (dermal or subcutaneous) soft tissue. This differentiation may be simple, but it becomes more difficult as the more aggressive and underappreciated inherently benign lesions are considered. Particularly difficult is myositis ossificans. The patient often has a history of trauma and often presents with a large, firm-to-hard lesion that, on plain film, may have intrinsic calcification. However, these signs do not preclude a malignant lesion. With myositis ossificans, Tru-Cut (CareFusion Corporation, San Diego, CA) needle biopsy or open biopsy is often accompanied by aggressive hemorrhage, which suggests a vascular neoplasm. In most cases, diagnosis can be made fairly accurately by either plain film or MRI scan. Certainly, myositis ossificans should be suspected when there is a significant history of trauma and the lesion is particularly hard and has inherent calcification.
For diagnosis to be accurate, the biopsy must be adequate and representative of the tumor, and the tissue must be well fixed and well stained. Antibodies for immunohistochemical staining are available commercially, and this technique is readily applicable to paraffin-embedded tissues. The most useful immunohistochemical markers are the intermediate filaments (e.g., vimentin, keratin, desmin), leukocyte common antigen, and S-100. In addition, the pathologist should be prepared to process tissue from selected cases for electron microscopy, cytogenetic studies, or molecular analysis. This requires that the clinician and pathologist communicate before the biopsy is performed to ensure that the necessary steps are taken in handling the tissue.
Cytogenetic analyses reveal specific clonal chromosomal aberrations, most commonly reciprocal translocations, in the majority of sarcomas.225,245–248 Among these are 11 different translocations involving the EWSR1 gene or its family members (FUS, TAF15) found in five different sarcomas. In a significant subset of sarcomas, translocations can be diagnostically and occasionally prognostically useful. Because conventional cytogenetic analysis is labor intensive and requires short-term culture of the sarcoma cells, molecular genetic techniques (e.g., reverse-transcriptase polymerase chain reaction and fluorescence in situ hybridization [FISH]) may be useful diagnostic adjuncts, particularly for diagnosing and distinguishing among the small cell sarcomas. Oligonucleotide and complementary DNA arrays may eventually add to the sophistication of determining the diagnosis and prognosis of such tumors.248 FISH testing for specific chromosomal abnormalities is now feasible for routine diagnostic use. FISH is also useful to identify supernumerary ring chromosomes, seen in mesenchymal neoplasms of low or borderline malignancy, such as dermatofibrosarcoma protuberans. Table 89.1 in Chapter 89 (“Molecular Biology of Sarcomas”) describes some of the genetic changes identified in soft tissue sarcomas.
As might be expected, there can be considerable disagreement among pathologists on the specific histologic diagnosis in individual cases. When a panel of expert pathologists reviewed pathologic material from 424 patients who entered into Eastern Cooperative Oncology Group sarcoma trials, 10% of cases were rejected as not being sarcoma, and 16% were the subject of disagreement on the histologic subtype.249 In the Scandinavian Sarcoma Group experience, the specific histologic diagnosis was disputed in 20% of cases.235 Of 1,463 histologic specimens obtained from patients with connective tissue tumors in France and Italy, grade and histologic subtype were confirmed by an expert pathologist in only 56% of cases. The discordance for 35% of patients was a different characterization of histologic subtype or grade, and in 8% was complete discordance (recharacterized as benign, different histology, or not a sarcoma).250 With increasing familiarity with the immunohistochemical and genetic studies needed to diagnose soft tissue sarcoma, the rate of this discordance may be decreasing.
Imaging
Imaging studies for soft tissue sarcoma vary, depending to some extent on the site. They involve evaluation of both the primary lesion and the potential site of metastasis. Evaluation of the primary lesion in the extremity and head and neck predominantly is by either CT or MRI. Although MRI provides some increased definition, a Radiology Diagnostic Oncology Group study comparing these modalities showed no benefit of MRI over CT.251 For the primary sarcoma of intra-abdominal, chest, or retroperitoneal locations, a spiral CT scan is preferable to MRI because air–tissue interface and motion artifacts often degrade MRI quality. In addition, spiral CT allows both the primary and potential for metastasis to be assessed in a single study. What is clear in this era of cost containment is that imaging with multiple modalities, all focusing on the same entity, is not required.
Positron Emission Tomography
Positron emission tomography (PET) has a number of potential uses in sarcoma management, although it has yet to gain universal acceptance. The standard uptake value in 18F-fluorodeoxyglucose (FDG) PET has been associated with histopathologic grade, cellularity, mitotic activity, MIB-1 labeling index, and TP53 overexpression, but it has never been proven to provide independent prognostic information and frequently fails to distinguish benign tumors from low-grade sarcomas.252–254
PET may also become useful for determining early responses to systemic therapy for soft tissue sarcoma. Specifically for primary extremity sarcomas, response by FDG-PET was superior to radiologic tumor size changes as a predictor of outcome after treatment with neoadjuvant chemotherapy.255–258 Similar results have been found for pediatric sarcomas. FDG-PET has been used for the early prediction of chemosensitivity in patients with soft tissue sarcomas who received neoadjuvant chemotherapy; the accuracy was 83% and positive predictive value for responders was 92%.259 Further studies are needed to determine if FDG-PET is sufficiently specific and accurate in determining chemotherapy response to alter clinical decisions on continuing, discontinuing, or modifying the treatment.
The current role of PET seems to be primarily in the identification of unsuspected sites of metastasis in patients with recurrent high-grade tumors, given the high rate of metastatic disease in this setting.
Imaging Sites of Metastasis
As important as imaging studies of the primary lesion is evaluation of possible sites of metastasis. For patients with extremity lesions, most metastases (70%) go to the lung.260 For patients with retroperitoneal or visceral lesions, a much more common site for metastases is the liver, with lung only a secondary site. Nevertheless, no site is immune from soft tissue sarcoma metastasis, and other patterns can be identified (e.g., intra-abdominal soft tissue metastases or pelvic or spinal bony metastases of extremity myxoid or round cell liposarcomas).122,261
Lymph node metastases are uncommon, except for certain histologic types usually associated with childhood sarcoma.262 In the MSKCC experience,262 3.7% of 1,066 patients with extremity soft tissue sarcoma had lymph node metastasis. Higher prevalence was seen in epithelioid sarcoma, 3 of 15; rhabdomyosarcoma, 4 of 21; clear cell sarcoma, 2 of 18; and angiosarcoma, 2 of 18. These findings were confirmed by a report from the Royal Marsden Hospital,263 with 3.4% of 2,127 patients having regional lymph node metastasis. Again, prevalence was higher with rhabdomyosarcoma, epithelioid sarcoma, and angiosarcoma. Thus, for these subtypes, the draining lymph node basin should be subjected to careful clinical examination and complete imaging if indicated.
Patients with visceral and retroperitoneal lesions should have their liver imaged as part of the initial abdominal CT or MRI. For extremity sarcomas, imaging for metastasis is less important for patients with low-grade or small superficial high-grade extremity lesions, and simple chest radiography will suffice. Conversely, for patients with deep or large high-grade extremity lesions, for which the risk of metastatic disease is significant, more extensive evaluation with a CT scan of the chest is often preferred. Although CT is the most commonly used modality to evaluate pulmonary metastases, it is more expensive than radiographs, delivers a higher radiation dose, and may give false-positive results because of small, indeterminate pulmonary nodules. One study correlated thoracotomy with CT and found that only 60% of malignant nodules <6 mm in size were found at thoracotomy.264 It is unclear if there is a better imaging modality to evaluate metastases of <1 cm. Newer techniques, such as FDG-PET, are being used to evaluate distant metastases and, when combined with CT and conventional imaging, may improve the diagnostic accuracy of preoperative staging. However, overstaging remains a problem in 12% of patients, and PET-CT remains limited in evaluating pulmonary metastases of <1 cm.265 FDG-PET lacks specificity in its ability to distinguish between low-grade malignancies and benign entities. An additional concern is that many low-grade sarcoma types and several high-grade types, like round cell liposarcoma, do not reliably show uptake for FDG, further limiting its routine use for staging sarcoma patients.
Biopsy
Biopsy can be used to evaluate malignancy, histologic grade, and sometimes histologic type. Precise knowledge of these features enables the treating physician to tailor the treatment plan to the tumor’s predicted pattern of local growth, risk of metastasis, and likely sites of distant spread. Either an incisional biopsy or several Tru-Cut core biopsies are required to obtain enough tissue for definitive diagnosis and accurate grading. The biopsy incision or core track should be placed in a location that can be completely excised at the time of definitive resection with minimal sacrifice of overlying skin. Excisional biopsy should be avoided, especially for lesions >3 cm in size, as the contamination of surrounding tissue planes may require the definitive resection to be more extensive.
In general, the important issue with biopsy is the adequacy of the sample. Sufficient viable tissue is required that is both representative of the lesion and available for histopathologic evaluation, immunohistochemistry, and, when necessary, cytogenetics and electron microscopy. As molecular markers become a factor in diagnosis, meticulous attention to the adequacy of biopsy, tissue preservation, and evaluation will be paramount.
Value of Tru-Cut Biopsy
Several studies have examined the value of Tru-Cut biopsy.266 Its accuracy is lower than for incisional biopsy, though substantially higher than for frozen section, and Tru-Cut biopsy has the advantage that it can be done in an office setting.
Fine Needle Aspiration Cytology
Fine needle aspiration (FNA) cytology has been examined by a number of authors but is usually used only for the confirmation of recurrence rather than for the primary diagnosis. Particular problems with FNA are the limited sampling and lack of tissue architecture, which degrade diagnostic accuracy. In addition, the amount of tissue collected usually does not allow for ancillary molecular diagnostic techniques.
Some authors have argued that biopsy itself is not justified if FNA is available. Rydholm267 suggested that open biopsy is never indicated, arguing that open biopsy risks local tumor spread and increases both the magnitude of the subsequent operation and the need for adjuvant radiation therapy. Using FNA, the surgeon proceeds directly to open operation. However, this requires referral before antecedent biopsy, a relatively uncontrollable event in the United States. Other authors suggest that this approach results in the referral of 10 patients with benign lesions for every sarcoma patient, certainly an untenable situation under our care system.
The no-biopsy approach presupposes that all that is required is a malignant sarcoma diagnosis and that the type or grade of sarcoma does not determine therapy. The use of FNA in patients with large sarcomas who are candidates for neoadjuvant therapy is also problematic due to difficulty in grading and subtyping these tumors accurately from such small samples. However, proponents argue that immunohistochemistry, electron microscopy, DNA cytology, and chromosomal analysis, all of which can be performed on FNA specimens, will ensure the appropriateness of this approach. However, the authors still favor obtaining adequate tissue from several Tru-Cut cores or an incisional biopsy to determine a definitive histologic diagnosis and grade before initiating treatment.
Frozen Section
In some institutions, frozen section is the diagnostic tool of choice. For diagnosis of malignancy, frozen section is accurate, but for histopathologic subtypes and grade, it is inferior to permanent sections of either Tru-Cut or incisional biopsy.266
Sarcoma Staging
The intent of staging systems is to group patients according to their probability of metastasis, disease-specific survival, or overall survival. The major staging used for soft tissue sarcoma is the system developed by the American Joint Committee on Cancer (AJCC). This system, first developed in 1992, has undergone significant changes, based on both histologic and clinical information. The current 2010 AJCC TNM (tumor, node, metastasis) system (Table 90.2) incorporates histologic type, histologic grade, tumor size, depth, regional lymph node involvement, and distant metastasis. It incorporates four major changes compared with the 2002 system. First, it excludes four histologic types: gastrointestinal stromal tumor (GIST), desmoid tumor, Kaposi sarcoma, and infantile fibrosarcoma. Second, it adds three histologic types: angiosarcoma, extraskeletal Ewing sarcoma, and dermatofibrosarcoma protuberans. Third, it reclassifies N1 disease from stage IV to stage III. Fourth, grading has changed from a four-grade to a three-grade system, although it accommodates two-, three-, and four-tiered grading systems.268
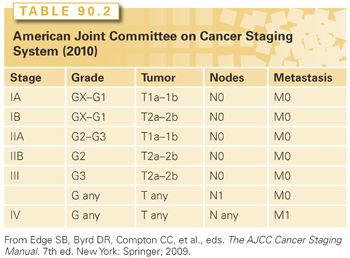
Analysis of the primary extremity soft tissue sarcomas seen at MSKCC during 1982 to 2009 suggests that the AJCC 2010 staging system nicely discriminates the probability of metastasis and disease-specific survival (Fig. 90.3 and Table 90.2). However, the AJCC staging still has limitations. A recent analysis suggested that risk assessment would be improved by increasing the number of primary tumor size categories from two to four. The analysis also suggested that patients with N1 M0 disease, whom the AJCC 2010 has moved to stage III, in fact have survival intermediate between that of other stage III patients and the stage IV patients.269 Unfortunately, this system still fails to adequately account for sarcomas located in the retroperitoneum and may incompletely account for the influence of specific histologic subtypes on outcome. There is as yet no adequate staging system for retroperitoneal or intra-abdominal lesions.
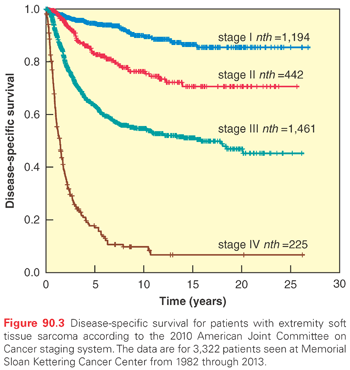
Nomograms
The accuracy of predicting patient outcomes can be increased by integrating multiple clinical and histologic features in a predictive model such as a nomogram. To predict sarcoma-specific survival, MSKCC researchers developed nomograms that integrate information on patient age and tumor size, grade (low versus high), depth, site, and histopathology. Nomograms are available for both primary lesions270 and locally recurrent271 lesions. These nomograms can be readily transferred to handheld personal organizers for instant calculation of disease-specific survival probability. Because no current grading system performs well for every histologic type of sarcoma, the authors’ groups have recently developed histology-specific nomograms in liposarcoma,37 synovial sarcoma,272 and GIST.273 Site-specific retroperitoneal nomograms have been developed274–276 as well as a local recurrence nomogram for patients with primary extremity sarcoma to improve selection for adjuvant radiation.277 As new molecular and genetic biomarkers are discovered and shown to have prognostic value, they can be incorporated into nomograms along with conventional clinical-pathologic variables. This would enable the treating physician to design a treatment strategy tailored to an individual patient’s risk of relapse and potential for an aggressive clinical course.
Histologic and Prognostic Factors for Primary Extremity and Truncal Sarcoma
Histopathology is related to anatomic site (see Fig. 90.1), and the common histologic types in the extremities and trunk are liposarcoma, myxofibrosarcoma, UPS/PMFH, and synovial sarcoma. Clinical and pathologic factors that influence outcome were determined from analysis of prospectively collected data from 1,041 patients older than 16 years with localized soft tissue sarcoma of the extremity.239 The 5-year survival rate was 76%, with a median follow-up of 4 years. Factors that increased risk of local recurrence were age, recurrent disease at presentation, positive margin, and fibrosarcoma or MPNST histology. Factors that increased the risk of disease-specific death were tumor size, depth, and grade, recurrent presentation, positive margin, and MPNST or LMS histology.
Distant recurrence was associated with tumor size, depth, and grade, recurrent presentation, LMS histology, and, to a lesser extent, any nonliposarcoma histology. High-grade lesions have a much greater risk of developing a distant metastasis in the first 30 months. Even low-grade lesions, however, have a slow but inexorable increase in risk of metastasis over the long term.239 Prognostic factors clearly vary with time. Grade is a dominant factor in early metastasis, but in late recurrence initial size becomes equally important.278 Postmetastasis survival for most patients is independent of factors involved in the primary presentation, although an association has been found with tumor size.
Bone invasion and neurovascular invasion have historically been considered bad prognostic features. However, bone invasion is relatively uncommon in soft tissue sarcoma, so it has not been uniformly included in any staging system.
Five-year survival does not guarantee cure. An analysis of patients disease-free 5 years after the diagnosis and treatment of extremity lesions showed that 9% would go on to have a recurrence in the next 5 years.279 Unfortunately, survival has not measurably improved with time when corrected for stage.280 A review of 1,261 completely resected extremity lesions by 5-year increments for 1982 to 2001 suggested that disease-specific actuarial 5-year survival remained unchanged (approximately 79%) over 20 years. For high-risk patients (those with high-grade, deep tumors of >10 cm), disease-specific survival remains at around 50%.
Site of disease is a clear determinant of outcome and an important prognostic factor. Patients with extremity and superficial trunk lesions certainly do better than patients with retroperitoneal and visceral sarcomas (Fig. 90.4). Death from local recurrence is uncommon in those with extremity lesions but occurs frequently in patients with retroperitoneal liposarcoma.
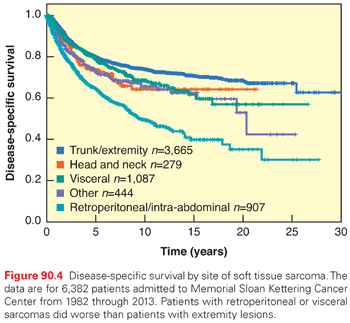
Innumerable molecular markers have been defined for soft tissue sarcoma—some with prognostic implications—but they have not been included in staging systems. However, the authors expect them to become increasingly important variables.
Diagnostic and Prognostic Factors for Primary Retroperitoneal/Intra-Abdominal Sarcoma
Most patients with retroperitoneal or intra-abdominal sarcoma present with an asymptomatic abdominal mass. On occasion pain is present, and less common symptoms include GI bleeding, incomplete obstruction, and neurologic symptoms related to retroperitoneal invasion or pressure on neurovascular structures. In one report, only 27% of patients with retroperitoneal sarcoma had neurologic symptoms, which primarily were related to an expanding retroperitoneal mass.281 Weight loss is uncommon, and incidental diagnosis is the norm. The diagnosis is usually suspected on finding a soft tissue mass on abdominal CT or MRI. Often the diagnosis is clear without biopsy, and many proceed directly to operative resection in the absence of a clinical trial. FNA biopsy or CT-guided core biopsy has a limited role in the routine diagnostic evaluation of these patients. CT-guided core biopsy is indicated if abdominal lymphoma, germ cell tumor, or carcinoma is strongly suspected. Preoperative biopsy is also indicated for patients who present with distant metastasis or advanced local disease that on abdominal or pelvic imaging appears to be difficult to completely remove surgically without substantial morbidity. In most patients, exploratory laparotomy should be performed and the diagnosis made at operation, unless (1) the patient’s tumor is clearly unresectable, (2) neoadjuvant chemotherapy or radiotherapy is needed to attempt to make the tumor more resectable, or (3) the patient will be undergoing preoperative investigational treatment.
CT remains the primary modality for evaluation of retroperitoneal and visceral sarcomas. Because the most likely site of visceral metastasis is the liver followed by lung, a CT scan of the chest, abdomen, and pelvis encompasses the primary lesion and the most likely sites of metastasis in a single examination.
Retroperitoneal or intra-abdominal sarcomas (excluding visceral sarcomas such as GIST) account for approximately 15% of all soft tissue sarcomas. The most common histologic types in the retroperitoneum are liposarcoma (60%), LMS (22%), solitary fibrous tumor (5%), and MPNST (2%). Among primary retroperitoneal liposarcomas (excluding pleomorphic liposarcoma and liposarcoma not otherwise specified), about 50% are low grade, with 46% and 3% classified as well differentiated and myxoid, respectively, and 50% are high grade with 49% and 2% classified as dedifferentiated and round cell liposarcoma, respectively.
An analysis of 278 patients with primary retroperitoneal sarcoma282 showed that grade and completeness of resection were the most important independent prognostic factors for disease-specific survival, with incompletely resected patients having survival similar to that of patients whose disease was unresectable from the outset. In this same study, histology and grade were both significantly associated with local recurrence, with liposarcoma having a 2.6-fold greater risk of local recurrence compared to other histologic types. Other studies have shown that for patients with retroperitoneal or intra-abdominal sarcomas, similar to patients with extremity sarcomas, the histologic type is an important prognostic factor for disease-specific survival, with this outcome worse for patients with MPNST and patients with LMS than for patients with solitary fibrous tumor (Fig. 90.5). Liposarcoma subtype provides additional prognostic information for disease-specific survival (see Fig. 90.5). In a multivariate analysis of liposarcoma, histologic subtype, completeness of resection, age, and contiguous organ resection were all independent predictors of disease-specific survival, with hazard ratios of 6 for subtype and 3.8 for completeness of resection.40
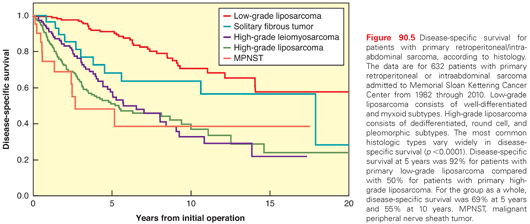
MANAGEMENT BY PRESENTATION STATUS, EXTENT OF DISEASE, AND ANATOMIC LOCATION
Management of Extremity and Truncal Sarcoma
Surgical Management of Primary Localized Disease
Although surgery remains the principal therapeutic modality in soft tissue sarcoma, the extent of surgery required, along with the optimum combination of radiotherapy and chemotherapy, remains controversial. The individual patient’s clinical and pathologic characteristics—particularly the pattern of spread expected for the patient’s histologic subtype—should be used to design the most effective treatment plan. Figure 90.6 shows a suggested algorithm for management of patients with extremity or truncal disease.
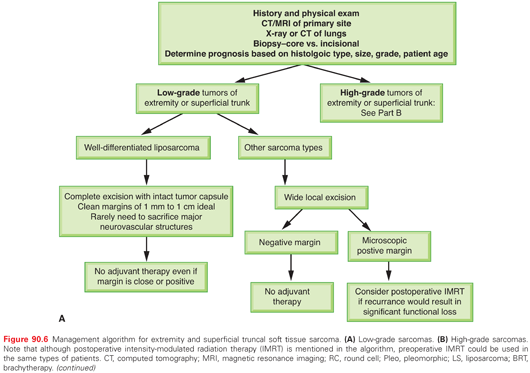
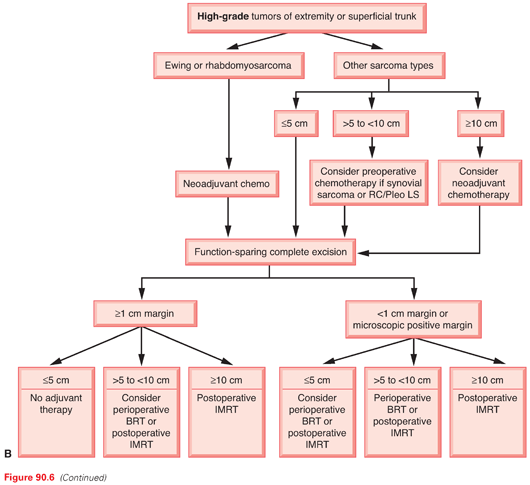
Extent of Surgical Resection. The most extensive resection, amputation, should be only rarely indicated for soft tissue sarcoma. At MSKCC, the amputation rate, which was 50% in the late 1960s, is now <5%. Outcomes from amputation versus limb-sparing surgery for extremity lesions were addressed by a prospective, randomized trial with well over 10 years’ follow-up. Although local recurrence is greater in those undergoing limb-sparing operation plus irradiation than in those undergoing amputation, disease-free survival is not different.283 Moreover, the level of handicap can be significantly lower in patients treated with limb-sparing surgery.284 Amputation should be reserved for tumors that cannot be resected by any other means, in patients without evidence of metastatic disease and with potential for good long-term functional rehabilitation.
For these reasons, surgery for extremity or truncal tumors most often consists of wide en bloc resection. Historical attempts to resect all muscle bundles from origin to exertion have been supplanted by an encompassing resection, aiming to obtain a 1-cm margin of uninvolved tissue in all directions. Two-centimeter margins are employed for histologic subtypes with infiltrative borders (e.g., DFSP or myxofibrosarcoma). For certain low-grade histologic types, however, even 1-cm margins are not required for excellent local control. For example, well-differentiated liposarcomas of the extremities require only complete excision with a minimal surrounding margin, as the majority of these tumors will not recur, even after a limited or microscopically positive margin excision, as long as the excision is complete. Skin surrounding the biopsy site, tethered to the tumor, or showing neovascularization in association with an underlying lesion should be removed with the specimen; myocutaneous flaps may be considered when a significant defect results or adjuvant radiation will be required. The limiting factor in obtaining wide margins is usually neurovascular or, occasionally, bony juxtaposition. Because very few soft tissue sarcomas invade bone directly, bone rarely needs to be resected; periosteum can be removed to provide an adequate margin when soft tissue sarcoma abuts the bone. Similarly, perineurium can be removed with the tumor to provide margins when the tumor is directly adjacent to a major motor nerve. In rare instances, a major nerve or vascular bundle is encased by a soft tissue sarcoma. Low-grade lesions may be bivalved to preserve the nerve; however, in the case of a high-grade tumor, resection may be required. Vascular bypass can be performed for arterial resections. Bracing can be used to compensate for removal of femoral or sciatic nerves.
Indications for Adjuvant Therapy. As detailed in the next section, radiation therapy should be added to limb-sparing surgery for some high-risk patients. Neoadjuvant chemotherapy or investigational approaches should also be considered for patients with high-grade lesions >10 cm and for those with synovial sarcoma or myxoid/round cell liposarcoma >5 cm (subtypes highly responsive to chemotherapy) (see “Chemotherapy for Primary Localized Extremity/Truncal Sarcoma”).
A significant subset of subcutaneous and intramuscular sarcomas can be treated by wide excision alone, with a local recurrence rate of only 8% to 20%.285–287 For example, in patients with small lesions (<5 cm), complete surgical excision with margins of >1 cm is usually sufficient without the need for any adjuvant therapy. Indeed, in a group of 159 patients with small primary tumors resected with negative margins, adjuvant radiotherapy showed no benefit: the 5-year local control rate was 77% in those selected to receive adjuvant radiation, compared to 92% in those undergoing surgery alone (p = 0.08). If, however, the margin is positive, the authors’ policy is to administer radiotherapy to patients with high-grade tumors even if the tumor was ≤5 cm. In contrast, patients with large, low-grade lesions such as atypical lipomatous tumors rarely require radiation therapy, as local recurrence rates are low (<10%) in patients treated with surgery alone.119
Another factor that should be considered is whether the patient has had a prior unplanned excision (i.e., excision without adequate preoperative staging or consideration of the need to remove normal tissue around tumor). At Princess Margaret Hospital, the rate of local recurrence was significantly higher in patients who were treated after unplanned excision on the outside than in patients who received their treatment at the institution (22% versus 7%; p = 0.03).288 Unplanned excision is very common in community settings when small soft tissue lesions are excised under the presumption that they are benign. In these cases, the authors attempt a re-excision if at all feasible; otherwise, patients are strongly considered for adjuvant irradiation.
Radiation Therapy for Primary Localized Extremity or Truncal Sarcoma
The goals of adjuvant radiotherapy in the management of soft tissue sarcoma are to enhance local control, preserve function, and achieve acceptable cosmesis by contributing to tissue preservation. Adjuvant radiation should be added to the surgical resection for most deep, large (>5 cm), high-grade sarcomas if the excision margin is close, particularly with extramuscular involvement, or if a local recurrence would necessitate amputation or the sacrifice of a major neurovascular bundle.289,290 Superficial lesions and smaller contained lesions confined to individual muscles may be managed with surgery alone in expert hands.257,291 For most other situations, however, evidence strongly suggests that surgery that does not achieve wide clearance through normal tissue has a significantly higher rate of local failure, and even some small lesions may behave adversely.
That adjuvant radiotherapy enhances local control with conservative surgical resection in soft tissue sarcoma overall has been demonstrated in two randomized clinical trials, one using external beam radiation therapy (EBRT) and the other using brachytherapy (BRT).289,290 However, whether the addition of radiotherapy confers a benefit for small (<5 cm) high-grade lesions is controversial.292
The contemporary era has brought newer techniques for radiotherapy planning and delivery that permit unprecedented accuracy in the use of both BRT and EBRT.
External Beam Radiation Therapy. EBRT is the most popular adjuvant radiotherapy approach, perhaps because EBRT relies less than does BRT on special technical and operational requirements. Nevertheless, EBRT requires comprehensive and multidisciplinary pretreatment consultation and accurate pathologic and radiologic assessment.
Postoperative Versus Preoperative External Beam Radiation Therapy. Postoperative EBRT was the first and remains the most widely practiced local adjuvant approach, in part because it does not require that surgery be postponed and because it allows for sterilization of microscopic nests of residual disease. Its use is supported by numerous single-institution studies and by a randomized trial that showed better local control from conservative surgery followed by EBRT than from surgery alone.290 Although the postoperative EBRT resulted in significantly worse limb strength, edema, and range of motion, these deficits were often transient and had few measurable effects on activities of daily life or global quality of life.290 Preoperative EBRT, however, has not been subjected to a randomized comparison against surgery alone.
Both preoperative and postoperative EBRT have their advantages and disadvantages. An advantage of postoperative EBRT is that the entire pathology specimen and final margins are available for pathologic analysis, helping to determine the need for further therapy. A major limitation is that the target is less precisely defined, and therefore volume is larger and dose is higher, resulting in greater late tissue morbidity. With preoperative EBRT, on the other hand, not only is the treatment volume well defined, but the blood supply is intact. The intact vascular supply may reduce the fraction of hypoxic cells particularly at the tumor margins, which under hypoxic conditions tend to be radioresistant, and thus may decrease the dose needed compared to postoperative radiotherapy. Confirmation of this principle, however, remains elusive. The major drawback of preoperative radiotherapy, as detailed subsequently, is that irradiation increases the risk of acute wound complications upon surgery. Which approach is superior remains unclear.
In a trial assessing EBRT sequencing, the Canadian Sarcoma Group randomized 190 patients with extremity soft tissue sarcoma to preoperative or postoperative radiotherapy. Preoperative radiotherapy doubled the risk of early acute wound complication, although this observation seems to apply almost exclusively to lower limb lesions.293 In the 5-year results from this trial,294 the preoperative and postoperative arms were nearly identical for local control (93% versus 92%, respectively) and metastatic relapse–free survival (67% versus 69%). In addition, the preoperative and postoperative arms did not differ significantly in overall survival (73% versus 67%; p = 0.5) or cause-specific survival (78% versus 73%; p = 0.6). Recently, a meta-analysis of pre- versus postoperative radiation in localized resectable soft tissue sarcoma suggested that the risk of local recurrence may be lower after preoperative radiation, and that the risk of metastatic spread is not increased with the delay in surgical resection necessary to complete preoperative radiotherapy.295
In the Canadian randomized trial, functional outcomes were collected prospectively.296 The trial design specified two validated instruments in addition an observer-based instrument.296 At 6 weeks after surgery, the preoperative group had inferior function, with worse bodily pain scores on all three rating instruments. However, at 3 to 12 months after surgery, the two groups showed no differences in these rating scores.296 It can be concluded that, for most of the first posttreatment year, the timing of radiotherapy has minimal effect on the function of patients with soft tissue sarcoma. The 2-year function and morbidity results297 show deteriorating late tissue sequelae (fibrosis and edema) in patients in the postoperative arm, resulting from larger radiotherapy doses and volumes. Of note, patients with significant fibrosis or edema had significantly lower function scores. In addition, patients who received the higher doses—most of them in the postoperative group—may eventually have a higher rate of bone fractures. These disadvantages of postoperative EBRT may override the higher frequency of acute wound complications with preoperative EBRT, although patients who do experience wound complications continue to experience some impaired function.298
Intensity-Modulated Radiation Therapy. The past decade witnessed an unprecedented improvement in delivery of EBRT to complex volumes and shapes. Leading these advances is intensity-modulated radiation therapy (IMRT),299 which uses computer algorithms for inverse planning and treatment delivery. IMRT may be particularly applicable to complex anatomic volumes such as retroperitoneal sarcoma juxtapositioned to liver or paraspinal lesions, for which conformal avoidance of liver or of kidney and spinal cord is necessary.300 Avoidance of bone is also possible,301,302 and the risk of radiation-induced fracture of a weight-bearing bone may be reduced by using specific IMRT dose avoidance objectives (see “Serious Complications of Primary Treatment”).
IMRT in the treatment of soft tissue sarcoma appears to enhance normal tissue protection while maintaining oncologic control. Notably, a recent phase 2 trial of preoperative IMRT in lower extremity sarcoma conducted at the Princess Margret Hospital indicated a 5-year local recurrence–free survival of 88% and very favorable doses to bone (mean dose 26.2 Gy, ± 8 Gy) and vulnerable soft tissues, in addition to absence of bone fractures and a reduction in the number and severity of wound complications.303 IMRT was associated with a similar 5-year local recurrence–free survival (92%) at MSKCC.304 The local control results for both IMRT studies were also very similar to those with conventional EBRT in both arms of the aforementioned Canadian trial that compared preoperative to postoperative radiotherapy.294 Taken together, these results indicate that any superiority of IMRT over conventional EBRT is not likely to be based on tumor control but on a reduction in acute and late toxicity. This especially applies to serious problems such as bone fracture.303,305 Wound complications may also be influenced by reduction in the volume of soft tissue irradiated, especially if the immediate tissues to be used for the wound closure can be spared.303
Volume Issues in External Beam Radiation Therapy. Given the generally favorable oncologic results after EBRT, a priority is ameliorating toxicity to normal tissue by exploring reduced treatment intensity, including administered doses or volumes. Prospective assessments of the volumes irradiated in EBRT are emerging, and these results will likely update the guidelines outlining optimal radiotherapy target volumes for EBRT.301–303
Targets are defined differently for preoperative versus postoperative radiotherapy. Preoperative radiotherapy can focus on the extent of definable disease (determined using imaging), and the target is based on the anatomic location, containment by barriers to spread (especially intact fascial planes), and allowance for geometric uncertainty related to potential variation in patient setup and physiologic movement. Special situations must also be considered, such as lesions arising in extracompartmental spaces such as the femoral triangle, antecubital space, and popliteal space, because these lesions have the ability to extend considerable distances proximally and distally with less anatomic restraint. Such lesions may spread to the neurovascular bundle early. The radiotherapy margins should reflect this and include undisturbed tissue planes and barriers to tumor incursion.
To cover any microscopic disease, the clinical target volume (CTV) for preoperative radiotherapy generally includes margins amounting to 4 cm longitudinally and 1.5 cm axially. This translates into field coverage approximately 5 cm long, allowing for beam penumbra and treatment uncertainties such as patient movement. Some controversy regarding planning of field coverage has persisted. A recent Radiation Therapy Oncology Group consensus panel, after reviewing data regarding the local extension of soft tissue sarcomas, set guidelines that included 3-cm longitudinal coverage and 1.5-cm radial coverage.306 However, these recommendations were not based on outcome efficacy results and conflict slightly with international recommendations provided by members of various cooperative clinical trials groups.307 The latter recommendations suggest that the 4-cm CTV longitudinal coverage should be maintained until contrary data emerge, as it is based on the only available prospective clinical trial data (the original Canadian preoperative versus postoperative trial), and because histologic assessment of adjacent tissues suggest that microscopic sarcoma cells may be present up to 4 cm away from the gross tumor volume.293,308 This longitudinal margin was also used in the recent Princess Margaret Hospital phase 2 prospective trial in lower extremity lesions, which is the only prosective outcome data with IMRT at this time.303
Postoperative radiotherapy volumes are significantly larger because they ideally encompass all surgically manipulated tissues, with the recognition that this is impossible in some anatomic sites due to the proximity of critical anatomy. In addition, because surgery has disrupted the anatomic planes and they no longer provide containment barriers to tumor growth, the entire CTV extending 4 cm beyond the surgically manipulated tissue must be considered high risk and thus treated for at least the first phase of irradiation (e.g., an appropriate dose is 50 Gy). Subsequently, the volume is reduced to the immediate tumor bed. Alternatively, with a single-phase IMRT approach, a higher dose could be delivered to the small high-risk volume while using a more moderate dose for the more peripheral regions of the CTV.
The international clinical trials group consensus panel mentioned earlier also generally recommended that the CTV extend 4 cm longitudinally beyond the surgically manipulated tissues.307 Currently, the volume of postoperative radiotherapy is being investigated in a randomized phase 3 trial conducted by the United Kingdom National Cancer Research Network. The trial compares a standard volume (5 cm longitudinal margin to gross tumor volume or 1 cm from the surgical scar, whichever is longer in the cranio–caudal direction and 2 cm axial margin) versus an experimental volume (2 cm longitudinal margin to gross tumor volume and 2 cm axial margin). Patients in both arms of the study will receive 66 Gy over 6.5 weeks; patients in the standard arm, but not the experimental arm, will have a volume reduction after 25 fractions (50 Gy). The goal is to assess if a reduced volume of postoperative radiotherapy will increase limb function without compromising local control.309
A final issue concerning choice of irradiation target volume concerns the definition of the areas that may harbor disease and especially whether the risk area should include any peripheral edema that surrounds the tumor. A study noted previously from the Princess Margaret Hospital, which correlated MRI characteristics with pathologic features, suggests that in some cases the edema does harbor sarcoma cells.308
Dose Issues in External Beam Radiation Therapy. The dose of radiotherapy represents an additional unexplored area. The preoperative dose used in most institutions is approximately 50 Gy in daily fractions of 1.8 to 2.0 Gy over approximately 5 weeks. Generally, a postoperative boost is administered only if the surgical margins are positive, and the benefit of this boost is unclear. For example, for patients who had positive surgical margins, Delaney et al.310 reported that the factors associated with local control included total radiotherapy dose >64 Gy (delivered as pre-, post-, or preoperative with a postoperative boost), with the implication that a boost is beneficial following preoperative radiotherapy. A different conclusion was drawn from a retrospective study at the Princess Margaret Hospital of patients with extremity soft tissue sarcoma and positive surgical margins. All patients had been treated with preoperative radiotherapy (50 Gy); 41 received a postoperative boost (typically 16 Gy) and 52 received no other radiotherapy. A common reason for omission of boost was wound complications, which tend to occur in more extensive and adverse cases. Despite this, patients who received a postoperative boost had worse 5-year estimated local recurrence–free survival (74% compared to 90% for preoperative radiotherapy only).311 This is consistent with recent pooled results from two Harvard groups, which showed no additional benefit to the radiotherapy boost in patients who had a positive surgical margin after preoperative radiotherapy and resection.312 Consequently at the Princess Margaret Hospital a postoperative boost is not used for patients with microscopically positive resection margins.
Adjuvant Brachytherapy. The fundamental interest in BRT stems from the ability to focus the dose directly on the tumor bed, thereby potentially improving the therapeutic ratio. Very high dose levels in the vicinity of the radioactive sources and the rapid dose fall-off permit dose intensification while protecting the surrounding normal tissue. Therefore, BRT usually spares more normal tissue than EBRT, except when precision techniques such as IMRT are used. Moreover, patients usually complete all their treatment in about 2 weeks compared to 6 to 7 weeks with EBRT, thereby limiting repopulation of residual tumor cells. The technical aspects of BRT differ from those of EBRT, and specific guidelines for its use and technical delivery have been published. BRT, unlike EBRT, requires a specific collaboration between surgical and radiation oncologists. With BRT, unlike in postoperative irradiation, no attempts are made to treat large margins or to include the scar and the drainage site, although it is acknowledged that this approach has not been formally compared with EBRT in similar cases.
The efficacy of BRT was compared to that of IMRT among 134 patients with primary high-grade extremity sarcoma in recent data from MSKCC.304 The BRT and IMRT groups were similar in terms of traditional prognostic factors, although the IMRT group had a significantly greater proportion of patients with positive or close margins, large tumors, and bone or nerve manipulation. The median follow-up was approximately 4 years. The 5-year local control rate was 92% for the IMRT group compared to 81% for the BRT group (p = 0.04). On multivariate analysis, IMRT was the only predictor of improved local control (p = 0.04).304 Earlier evidence from MSKCC suggests that BRT may not provide optimal results in regions unsuited for ideal implant geometry, such as in more proximal regions of the limb.292
In patients treated with BRT as the sole radiotherapy, the dose is usually 45 Gy given over 4 to 6 days, and when given as a boost, the dose is usually 15 to 20 Gy from BRT plus 45 to 50 Gy from EBRT. Of importance, the catheters are loaded no sooner than the sixth postoperative day to allow time for wound healing.313 The most commonly used isotope is low–dose-rate 192Ir; however, high-activity 125I is occasionally used in young patients or to protect the gonads. High–dose-rate 192Ir has been advocated to take advantage of its radiation safety and dose-optimization capabilities.
The BRT CTV may be difficult to define, but in general it is represented by the volume of tissue considered at risk for microscopic extension of tumor and includes the tumor bed visualized on radiographic studies and under direct inspection intraoperatively. The results with adjuvant BRT suggest that radiation treatment directed to the tumor bed plus a 2-cm margin is adequate.314 Although there is no consensus on the exact size of the margin beyond the tumor bed, generally at least 2 cm longitudinally and 1 cm axially are recommended.307
Related posts:



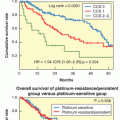

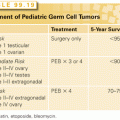
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


