Chapter 63 Soft Tissue Sarcoma
Soft tissue sarcomas (STS) are rare malignancies derived from mesenchymal tissue and comprise approximately 1% of adult cancers in the United States, resulting in almost 10,500 new cases annually; approximately 5700 occur in males and 4800 affect females.1,2 No obvious association with ethnicity seems evident. The disease is characterized by heterogeneity in anatomic site, histologic tumor subtype, and risk of metastatic progression. All age groups are affected, with pediatric sarcomas frequently occurring during infancy whereas the incidence of adult-type sarcomas increases in the final decades of life3; the median age at onset of STS is generally from 50 to 55 years. Just over half originate in the limbs (extremities), whereas approximately 40% arise in the retroperitoneum/visceral tissues and torso (truncal), with the remainder affecting the head and neck (10%).4 Because of different conventions in attribution of certain sarcomas to the abdomen and thorax there has been a recent increase in incidence in these sites.
Etiology and Epidemiology
Environmental Etiologic Factors
A perplexing issue about STS is that in most cases no clearly defined etiologic factor is evident. Notwithstanding this, several factors have been identified that either predispose or are associated with the development of STS. Potential risk factors need to be further assessed in STS such as herbicide exposure and constitutional and hormonal factors during childhood, puberty, and adulthood. STS were identified in a group of non–AIDS-defining neoplasms linked to various concurrent viral infections in the human immunodeficiency virus (HIV)-infected host, including children.5,6 No clear demographic features have been identified in normal populations, although one Italian study questioned whether body weight contributed to increased risk.7
The factor with perhaps the greatest notoriety is radiation resulting from accidental, therapeutic, or diagnostic exposure. Radiation-associated STS have demonstrated inferior survival compared with sporadic STS, even if one adjusts for known prognostic factors such as histologic type, size, age, margin status, and site.8 The secondary risk of cancer has been enshrined in the collective oncology consciousness from as long ago as 1922.9 However, certain conditions in themselves are associated with intrinsic host predisposition to radiation-induced neoplasia, most notably retinoblastoma. Although rare in the cancer incidence statistics, children are an obvious concern because of the available latent period that can span many decades.
Although the risk associations with RT are well appreciated, the molecular mechanisms and real antecedent cause of these lesions are poorly understood. Germline mutations in tumor suppressor genes and/or accumulated damage to DNA repair genes may lead to neoplasia, but it remains unknown if most individuals have a genetic defect that underpinned the development of the first cancer, for which RT was administered, and, in turn, led to the induction of the RT-induced cancer.10 If this is the case, there may be subgroups of individuals at considerably greater risk, whereas others have relatively low risk.10 One hypothesis is that incomplete damage in normal tissues results in mutagenic responses and disorganized reparative proliferation that can eventually trigger tumor induction.11 This may provide a basis for the observation that RT-induced sarcomas commonly originate on the perimeter of the previous RT target volume and not necessarily in the high-dose region.
RT use in the primary management of breast cancer remains a problem from the standpoint of sarcoma induction. Karlsson and colleagues12 studied 122,991 women with breast cancer in Sweden from 1958 to 1992 and found 116 STS (40 angiosarcomas and 76 other lesions). Mery and associates13 reported the risk factor for development of STS for 563,155 women with breast cancer who presented from 1973 to 2003. A total of 948 patients developed STS; patients who received RT had a higher incidence of all STS (1.5-fold), in particular angiosarcoma (9-fold) and malignant fibrous histiocytoma. The incidence peaked at 10 years and remained elevated up to 20 years after RT. These authors also found that patients who underwent partial mastectomies had a 7-fold higher risk of developing secondary angiosarcoma when compared with those who underwent total mastectomies, after adjusting for RT and lymph node dissection. Having breast cancer also may incur additional risk because lymphedema after the treatment of breast cancer is independently associated with the development of lymphangiosarcoma.14 In this setting, the lymphangiosarcoma generally develops beyond the immediate irradiated volume in addition to the area that was targeted.15
Trauma is a controversial factor. Often, a minor episode of injury may draw attention to a preexisting mass or awkward malignant masses may be more prone to trauma and consequent injury. Pukkala and collegues16 studied cancer incidences in world class athletes in Finland. Smoking-related cancers were less frequent, whereas the incidence of other cancers was not reduced. The authors speculated whether injuries during active sport may predispose to sarcoma, and, in turn, there has been concern that operative trauma, including arthroplasty, may increase the risk of STS. However, Scandinavian studies17,18 on more that 100,000 patients who had undergone total hip or knee arthroplasty showed no increased risk of sarcoma and there was no sarcoma case presenting at the site of operation. Desmoid tumors are commonly seen in the anterior abdominal wall after pregnancy, although the biologic and traumatic basis for this is unclear.19
Several chemical carcinogens, including thorotrast, vinyl chloride, and arsenic, are established in the causation of hepatic angiosarcomas. Workers exposed to phenoxyherbicides, chlorophenols, and dioxins had higher mortality rates from cancer compared with controls, and the increase was highest for STS.20–22 There have been conflicting reports about occupational exposure to phenoxyacetic acids detectable in chlorophenols (frequent in wood preservatives) and certain herbicides.23–26
Molecular Pathogenetic Mechanisms
Contemporary appreciation of the molecular pathogenesis and behavior of STS suggests that these lesions are divisible into two major genetic groups: (1) those possessing specific genetic alterations and usually simple karyotypes, such as reciprocal chromosomal translocations (e.g., SS18-SSX1 or SS18-SSX2 in synovial sarcoma); and (2) sarcomas with nonspecific genetic alterations and complex unbalanced karyotypes.27 However, these concepts are considerably more complex and are evolving rapidly,28 and only a brief discussion is presented here.
Simple Karyotypic Sarcomas with Specific Genetic Alterations
The first group, those characterized by specific recurrent chromosomal translocations, comprises approximately one third of all sarcomas, and the resulting product of the specific gene fusions usually encode aberrant chimeric transcription factors. These translocations are often the only cytogenetic abnormalities and appear to be pathogenetically important. However, although successful cloning of most genes involved in recurrent translocations has taken place, there remains a paucity of information about the downstream targets mediating oncogenic transformation. In general, the promiscuous pairing of a proximal gene that contributes a promoter and functional domain, with a distal gene possessing a DNA binding domain that confers target specificity, determines the phenotype of various STS.27 Most translocations result in the production of a tumor-specific RNA that encodes a novel transcription factor that plays a role in oncogenesis in mesenchymal cells.
One uncommon mechanism results in a chimeric autocrine growth factor and is evident in dermatofibrosarcoma protuberans (DFSP) in which the t(17;22)(q22;q13) translocation involving the COL1A1-PDGFB genes provides a therapeutic target for response to the tyrosine kinase inhibitor imatinib. Mechanisms such as this represent opportunities for future management. At this time, however, they are typically used in a diagnostic manner to identify a particular subtype of tumor. This is the case for synovial sarcomas, nearly all of which exhibit the t(X;18)(p11;q11) translocation and can be distinguished from other tumors with similar morphology, such as a malignant peripheral nerve sheath tumor. Another example is the fusion product PAX3-FOXO1 arising from the t(2;13)(q35;q14) translocation as well as the t(1;13)(p36;q14) translocation expressed in alveolar rhabdomyosarcoma27 (Table 63-1).
TABLE 63-1 Cytogenetic Abnormalities and Fusion Genes in Soft Tissue Sarcoma
| Histologic Subtype | Usual Translocations | Genes Involved |
|---|---|---|
| Alveolar sarcoma of soft parts | t(X;17)(p11;q25) | TFE3-ASPL |
| Angiomatoid fibrous histiocytoma | t(12;16)(q13;p11) | FUS-ATF1 |
| Clear cell sarcoma | t(12;22)(q13;q12) | EWS-ATF1 |
| Congenital fibrosarcoma | t(12;15)(p13;q25) | ETV6-NTRK3 |
| Dermatofibrosarcoma protuberans | t(17;22)(q22;q13) | COL1A1-PDGFB |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWS-WT1 |
| Endometrial stromal sarcoma | t(7;17)(p15;q21) | JAZF1-JJAZ1 |
| Ewing’s/peripheral primitive neuroectodermal tumor | t(11;22)(q24;q12) | EWS-FLI1 |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) | EWS-NR4A3 |
| Fibromyxoid sarcoma, low grade | t(7;16)(q33;p11) | FUS-CREB3L2 |
| Inflammatory myofibroblastic tumor | t(1;2)(q22;p23) | TPM3-ALK |
| Myxoid liposarcoma | t(12;16)(q13;p11) | FUS-DDIT3 |
| Rhabdomyosarcoma (alveolar) | t(2;13)(q35;q14)t(1;13)(p36,q14) | PAX3(or7)-FOXO1 |
| Synovial sarcoma | t(X;18)(p11;q11) | SS18-SSX1 or SSX2 |
Note: Gene nomenclature evolves at least as rapidly as pathologic terminology.
Modified from Antonescu CR: The role of genetic testing in soft tissue sarcoma, Histopathology 48:13-21, 2006.
Complex Karyotypic Sarcomas without Specific Genetic Alterations
The remaining two thirds of STS lack a recurrent genetic signature and are characterized by numerous aberrations, including chromosomal losses and gains. Most adult spindle cell tumors, leiomyosarcoma, and pleomorphic sarcomas belong to this group. At the molecular level, this sarcoma subset features a high prevalence of TP53 checkpoint alterations, including TP53 inactivating mutations, homozygous deletion of CDKN2A, MDM2 amplifications, and so on. Unfortunately, most cases with complex karyotypes and multiple aberrations do not have a consistent correlation with pathologic or clinical parameters. There also appears to be no indication in most of these lesions that detectable genetic alterations might be useful either for diagnosis or prognosis.29
Genetic Conditions Associated with Sarcomas
Given the genetic information just presented, it is not surprising that STS are associated with several genetic conditions. Patients with neurofibromatosis have a 5% to 10% lifetime risk of developing a malignant neurofibrosarcoma arising in a neurofibroma,30–33 most commonly in the central nervous system. These patients frequently present the additional diagnostic dilemma of distinguishing between potential metastases or other benign neurofibromas found on staging investigations.
A significant risk for STS is found in individuals with somatic mutations in the TP53 tumor suppressor gene, including Li-Fraumeni syndrome, which is characterized by a familial cluster of sarcoma (12%), breast cancer (25%), leukemia, adrenal carcinoma, brain tumors (12%), lung cancer, skin cancer, and pancreatic cancer.34,35
Long-term survivors of retinoblastoma with the associated RB gene abnormality often develop tumors later in life, with a risk of 10% to 15%.36,37 For hereditary retinoblastoma, the cumulative incidence of a second cancer at 50 years after diagnosis is 50% compared with only 5% for nonhereditary retinoblastoma.38 Wong and associates38 showed that the relative risk of STS increases with dose beginning at 5 Gy, rising to 10.7-fold at dose levels exceeding 60 Gy.
Gardner’s syndrome, a subset of familial adenomatous polyposis, is associated with the development of intra-abdominal desmoids.39
Biologic Characteristics and Molecular Biology
As already mentioned, sarcomas have extensive cytogenic abnormalities that may aid in their differential diagnosis and may ultimately provide therapeutic targets. Unfortunately, molecular prognostication and prediction of treatment response are proving more elusive than was originally anticipated. For example, as previously mentioned, the presence of the translocation t(X;18)(p11;q11) has been used to confirm the diagnosis of synovial cell sarcoma for patients with poorly differentiated lesions that cannot be otherwise characterized. Irrespective of their histologic appearance, almost all synovial sarcomas contain the t(X;18)(SS18-SSX) translocation involving the more prevalent SSX1 (associated with monophasic histology)40 or SSX2 (associated with biphasic histology) gene, two closely related genes from chromosome Xp11, and the SS18 gene from chromosome 18q11. The result is the formation of a chimeric gene thought to function by encoding a transcription-activating protein. It was suggested that the transcripts of these fusion genes had specific prognostic significance,41–43 but results have been contradictory.44
Tissue hypoxia appears associated with the development of distant metastases independently from depth, size, and grade.45 Expression of osteopontin (OPN), a phosphorylated glycoprotein, appears to be induced by tumor hypoxia.46,47 Elevated serum or plasma levels of OPN have been correlated with tumor progression and metastasis in a variety of cancers.48,49 One study based on tumor and serum samples from 93 adult patients with STS found that an elevated OPN protein level in serum and tumor is a significant negative prognostic factor. Elevated OPN levels were associated with higher stage, higher grade, subtype, larger tumor size, and an increased rate of relapse.50 In addition, elevated OPN levels have been associated with decreased overall survival (OS), suggesting its potential use as a prognostic marker.51 Presumably one mechanism could be represented by hypoxia-induced molecular aberrations linked to expression of OPN.
Pathology and Pathways of Spread
Pathology
Classic Pathology Classification
While originating from the mesoderm and in some lesions from ectoderm, a beguiling feature of the legacy of the nomenclature of sarcoma pathology is the traditional view of histologic subtypes originating from similar-appearing normal tissue counterparts. The most common classification scheme for STS is still based on histogenesis, as described by the World Health Organization (WHO) and others52,53,54 (Table 63-2). However, as the degree of histologic differentiation becomes less apparent, the determination of a putative cellular origin becomes increasingly difficult.
TABLE 63-2 Histologic Classification of Soft Tissue Sarcoma*
| Fibrous Tumors |
| Fibrohistiocytic Tumors |
| Lipomatous Tumors |
| Smooth Muscle Sarcomas |
| Skeletal Muscle Sarcomas |
| Malignant Tumors of Blood and Lymph Vessels |
| Malignant Perivascular Tumors |
| Malignant Synovial Tumors |
| Malignant giant cell tumor of tendon sheath |
| Malignant Neural Tumors |
| Paraganglionic Tumors |
| Extraskeletal Cartilaginous and Osseous Tumors |
| Pluripotential Malignant Mesenchymal Tumors |
* Benign lesions other than fibromatosis and atypical lipoma are not included in the present tabulation.
Adapted from references 52, 53, and 54.
This view of pathogenesis and histogenesis is now being contested. Although some benign lesions (especially lipomas) may resemble mature differentiated normal tissue, many STS arise in tissues that do not typically possess the putative normal tissues mentioned. For example, synovial sarcomas are only rarely found in synovial tissues. Because the necessary genetic information is contained in all diploid cells, a more plausible explanation may be that a given set of genes may program differentiation in any mesenchymal cell, thereby giving origin to almost any mesenchymal neoplasm.52,53,54 It seems unlikely that there is a single precursor primitive mesenchymal stem cell for each sarcoma.
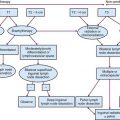
Classification by Functional Genomics
With advances in molecular characterization of STS, and the emerging dissatisfaction with traditional methods of classification, it is not unexpected that recent proposals have explored gene expression profiling for these tumors (Fig. 63-1) to attempt to identify a genome-based classification scheme.55,56,57,58,59,60 For example, Segal and colleagues59 examined RNA sarcoma samples and found that synovial sarcomas, round-cell/myxoid liposarcomas, clear cell sarcomas, and gastrointestinal stromal tumors (GIST) displayed distinct and homogeneous gene expression profiles. In a separate study, Nielsen and colleagues58 observed that KIT appeared to be one of the prominent discriminator genes for malignant fibrous histiocytoma. More recently, Nakayama and co-workers57 have shown that many neoplasms initially (mis)classified as undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma based on their morphology and immunoprofile could be reclassified as myxofibrosarcoma based on gene expression, suggesting that gene profiling will be a useful tool to aid histologic diagnosis of malignant fibrous histiocytoma. Two genes, GPR64 and TNXB, were particularly expressed by myxofibrosarcomas but not by undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma, thus allowing distinction between these two histotypes.
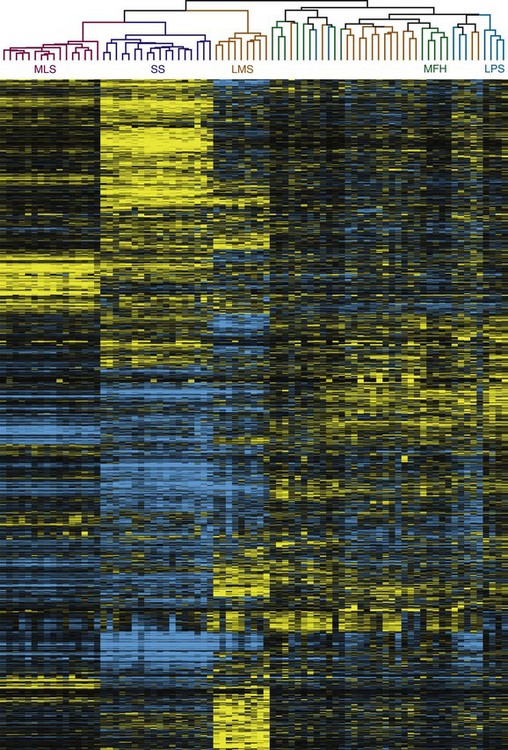
New molecular technologies such as comparative genomic hybridization array–based techniques and gene expression analyses show promise for the identification of genes, gene products, and signaling pathways involved in the pathogenesis, progression, and metastatic dissemination of sarcomas that may provide new strategies for the enhancement of local/systemic control and the reduction of the toxic effects of sarcoma management.56 In particular, nanotechnology offers countless opportunities for novel diagnostic and therapeutic approaches to selectively target tumors at the cellular level.60
Grading of Sarcoma
Several grading schemes have been proposed and validated as efficacious and are discussed in detail elsewhere.61 The three-tier system proposed by the French Federation of Cancer Centres is precisely defined, easy to use, and the most widely employed. The French system relies on a relatively balanced evaluation of parameters (differentiation score, mitoses, necrosis), but its greatest limitation lies in the assignment of a differentiation score. Roughly defined as the extent to which a lesion resembles normal tissue, differentiation score has little applicability for tumors that ostensibly have no normal tissue counterpart. It is also recognized that no system performs perfectly in all sarcomas. In one review, Deyrup and Weiss61 highlighted problems, including the fact that some STS do not lend themselves well to grading. These include (1) those in which grade provides no additional information (e.g., well-differentiated liposarcoma/atypical lipomatous neoplasm, Ewing’s sarcoma); (2) “ungradable” histologic subtypes (e.g., epithelioid sarcoma, clear cell sarcoma, angiosarcoma); and (3) sarcomas that have traditionally been graded but where this characteristic does not appear to differentiate these lesions prognostically (e.g., malignant peripheral nerve sheath tumor).
In addition to the French system, there is a U.S. National Cancer Institute (NCI) system.62 Both use degrees of necrosis and mitotic rate, but the American system considers histologic type or subtype, location, cellularity, and nuclear pleomorphism. The situation is further complicated by the potential use of different tiers of grade within existing classification systems (e.g., two-, three- and four-grade classification systems). The TNM stage classification of the American Joint Committee on Cancer (AJCC) now employs a three-tiered grading system based largely on the wish to incorporate the French system.63
Pathologic Features and Natural History
Selected Pathologic Subtypes
Malignant Fibrous Histiocytoma
Since the late 1970s malignant fibrous histiocytoma has borne the mantle of most common STS of middle and late adulthood and therefore warrants specific mention in the chapter. Originally described in the 1960s, the term was used to describe a group of sarcomas deemed to be derived from a mixed histiocytic and fibroblastic lineage. The existence of ‘‘malignant fibrous histiocytoma’’ as a distinct entity is now considered controversial and at best is regarded as a heterogeneous group of tumors without a specific known line of differentiation. Most of these tumors were considered to be either a variable storiform and/or undifferentiated pleomorphic phenotype or one of four rarer additional types: myxoid (the next most common subtype), giant cell, inflammatory, and angiomatoid (a rare variant and the only one to survive contemporary reclassification). Reclassification of many tumors in this group has afforded better prognostication, and the old term “malignant fibrous histiocytoma” now is generally considered obsolete.54
Current nomenclature recognizes the entities of undifferentiated high-grade pleomorphic sarcoma (previously storiform-pleomorphic “malignant fibrous histiocytoma”) and myxofibrosarcoma (formerly myxoid “malignant fibrous histiocytoma”), which is now the most common sarcoma of older adults, peaking in incidence in the seventh and eighth decades and presenting most frequently in the lower extremity. Most of these tumors are deep seated, developing in fascia and skeletal muscle, and have a high potential for local recurrence.54,56 Low-grade myxofibrosarcoma is known to have a particularly infiltrative growth pattern, and imaging evaluation with particular attention to tail-like extensions should be considered for management.64 This type must be differentiated from other sarcomas containing myxoid areas, such as pleomorphic liposarcoma. The feature that sets malignant fibrous histiocytoma lesions apart is the absence of a defined line of differentiation from which epithelial, melanotic, and lymphoid differentiations have been excluded.52
The natural history is extraordinarily variable, reflecting the heterogeneity of these lesions. The most common site for metastases from pleomorphic sarcomas is the lung (90%), bone (8%), and liver (1%), with rare regional lymph node metastases.54
Angiosarcoma
Angiosarcoma is currently included in the seventh edition of the TNM stage classification.63 Particularly unique is the propensity of superficial angiosarcoma to occur in the dermal tissues of the head and neck manifesting typically on the scalp (about 50%) or facial skin. These neoplasms commonly present as purple, bruiselike lesions in elderly white men and are rarely seen in patients of African origin.65 The macules usually become nodular, may coalesce, and may ulcerate. Frank bleeding is an ongoing and major problem and often results in chronic oozing through dressings. A particularly difficult aspect of management is the apparent multifocal nature that makes judgment about the area of risk almost impossible from the standpoint of accurate definition of margins for both surgery or RT. The clinical examination must be meticulous because it is the only real means of identifying the deceptive areas of multifocal involvement that may exist.66 Frequently, individual patches coalesce into flat masses of substantial size. Involvement of the eyelid and periorbital tissues is particularly troublesome. Metastasis is common if patients survive sufficiently long and occurs most typically in regional lymph nodes and the lung.
The majority of angiosarcomas are considered to originate from the endothelium of the blood vasculature.67 Koch and colleagues68 have outlined the pathologic features, which can range from well to poorly differentiated lesions. They are typically composed of an accumulation of an irregular or sinusoidal pattern of vessel with vascular spaces lined by a single row of atypical endothelium that may be several layers thick. Highly cellular lesions can manifest as sheets of cells with vascular space obliteration that contribute solid components as vascular channels effaced by the crowding of cells. High-grade, poorly differentiated lesions can be composed of undifferentiated cells and disordered architecture, making them difficult to discern from other histologies, although the hallmark clinical presentation and behavior generally leave little doubt about the diagnosis. On immunohistochemistry, these tumors are usually positive for factor VIII–related antigen, vimentin, CD34, and CD31.
It is suggested that angiogenesis and vascular permeability play a central role in the development of angiosarcomas. This is likely influenced by vascular endothelial growth factor A (VEGF-A), which plays a significant role in angiogenesis and vascular permeability. VEGF-A induces angiogenesis by acting through a tyrosine kinase receptor predominantly found on vascular endothelial cells, including VEGF receptor-1. VEGF-A acts to enhance the growth of tumors by promoting a more extensive blood vessel supply and increases the probability of hematogenous metastases. Similarly, it has been proposed that angiogenesis and vascular permeability play a central role in development of angiosarcomas, suggesting a role for VEGF-A in the pathogenesis of angiosarcomas,67 thereby opening the potential for molecular targeted agents in the future. Bevacizumab, a humanized monoclonal antibody to VEGF, is an attractive agent to consider in angiosarcoma, given its ability to inhibit tumor growth. Koontz and coworkers69 have reported promising results in two patients using neoadjuvant bevacizumab combined with RT.
Rhabdomyosarcoma
Rhabdomyosarcoma is especially important because it is the fifth most common cancer in childhood,70 but it also occurs in the adult, where the outcome is significantly less favorable.71 One feature concerns the “spindle cell rhabdomyosarcoma” that is only rarely described in the adult compared with children and carries a relatively favorable prognosis. In the adult it appears to have a predilection for the head and neck and may be unusually aggressive.72,73 The rhabdomyosarcoma classification recognizes embryonal, botryoid, alveolar, and pleomorphic subtypes for both childhood and adult cases. Rhabdomyosarcoma can be recognized on light microscopy by the presence of cross striations within cytoplasmic fibrils of spindle-shaped cells that typically demonstrate immunostaining for myogenic markers.74 Seventy percent of rhabdomyosarcomas can be classified as embryonal and 20% alveolar, and the remainder are variants, including the uncommon pleomorphic subtype. The pleomorphic subtype is almost never seen in the pediatric population and is not considered in those classifications; there is debate about whether it truly represents part of the disease process and it may fall into the “malignant fibrous histiocytoma” type categories described earlier.
A diagnosis of alveolar rhabdomyosarcoma is made if the tumor has alveolar-like spaces that are filled with round malignant, eosinophilic cells. Presence of any amount of alveolar morphology in the lesion qualifies it to be alveolar rhabdomyosarcoma, and alveolar morphology has been identified as a poor prognosticator. There is also a solid variant of this tumor that lacks alveolar spaces but is still included in this subtype. The presence of sheets of anaplastic cells warrants a diagnosis of pleomorphic rhabdomyosarcoma. Today the diagnosis of the less favorable alveolar subtype is generally based on the identification of the characteristic chromosomal translocations t(2;13)(q35;q14) and t(1;13)(p36,q14) and their fusion products (see Table 63-1).27
Both translocations may be associated with different clinical phenotypes.75 The translocations and the gene fusion products are characteristic, and the PAX3 gene family involved in the disease fingerprint may have a role in muscle development.76,77
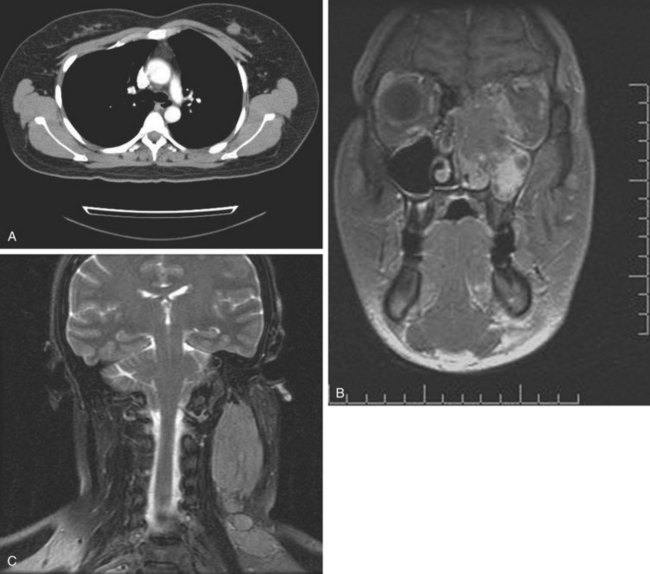
Rhabdomyosarcoma is also one of the sarcomas associated with a risk of lymph node metastasis. Irrespective of age, the alveolar and embryonal subtypes manifest high rates of response to chemotherapy and RT. The initial evaluation of rhabdomyosarcoma is similar to regular STS but should ordinarily include bone marrow examination. Sampling of cerebrospinal fluid should be considered in patients with parameningeal lesions. Rarely, but distinctly, there is a tendency for these lesions to develop synchronous or metachronous metastases in one or both breasts (Fig. 63-2).
Liposarcoma
Liposarcoma is the second most commonly encountered subtype of STS, and myxoid liposarcoma (MLS) is the most common variant.78 Classic MLS has a t(12:16) translocation, resulting in the TLS-CHOP fusion shared with the more aggressive round cell variant and not seen in predominantly myxoid, well-differentiated liposarcoma.79 It also has an unusual pattern of recurrence that includes a predilection for soft tissue metastases.80–83,84
Apparent multifocal presentation or recurrence at two or more anatomically separate soft tissue sites (with predilection for the retroperitoneum and mediastinum) and bone metastases are a striking yet unpredictable feature of myxoid liposarcoma and contrasts to other STS (Fig. 63-3). Surgery and RT may result in relatively long disease-free intervals (e.g., many years). Uncertainty had existed about whether such multifocal lesions represent an unusual pattern of metastasis or multiple separate primary tumors until Antonescu and associates85 showed that all cases of multifocal myxoid liposarcoma bore the identical molecular lineage of the parent primary tumor.
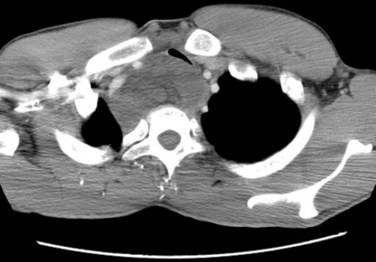
Figure 63-3 CT showing isolated mediastinal relapse with tracheal obstruction occurring in a patient who had presented 7 years previously with a myxoid liposarcoma in the popliteal fossa similar to the lesion shown in Figure 63-9. The initial limb lesion was treated with preoperative RT and maintained local control. This patient underwent airway management, preoperative RT, and surgical resection and was disease free with 15 months of follow-up. Isolated soft tissue relapse in this fashion is a hallmark of the behavior of myxoid liposarcoma and may result in long-term remission or salvage of such lesions.
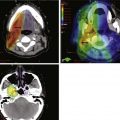
More favorable survival in a large cohort of liposarcoma patients compared with other histologic subtypes has been reported in a series exceeding 1000 cases. The observation was independent of other prognostic factors, including grade, depth, and tumor size.86 Myxoid liposarcoma also appears to manifest an unusually sensitive response to RT87,88 (Fig. 63-4). Two theories, which are not mutually exclusive, of the underlying mechanism have been postulated. RT might cause adipocyte-like maturation/differentiation of tumor cells,88

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


