Abbreviations
ACVR
activin A receptor
ALK
activin receptor–like kinase
AP1
activator protein 1
Bglap
bone gamma-carboxyglutamate protein
BMD
bone mineral density
BMP
bone morphogenetic protein
CCN
Cyr61, CTGF, Nov
CMV
cytomegalovirus
CTGF
connective tissue growth factor
CUB
clr/cls urchin endothelial growth factor BMP domain
Dmp
dentin matrix protein
ERK
extracellular regulated kinase
FGF
fibroblast growth factor
GH
growth hormone
HGF
hepatocyte growth factor
HIF
hypoxia-inducible factor
IGF
insulin-like growth factor
IGFBP
insulin-like growth factor binding protein
IL
interleukin
IRS
insulin receptor substrate
MAP
mitogen-activated protein
MMP
matrix metalloproteinase
Mt1
metallothionein 1
Nov
nephroblastoma overexpressed
PAPP-A
pregnancy-associated plasma protein-A
PDGF
platelet-derived growth factor
PDGFR
PDGF receptor
PI-3
phosphatidylinositol-3
PKC
protein kinase C
Prx1
paired homeobox gene enhancer
PTH
parathyroid hormone
RANKL
receptor activator of nuclear factor–κ-B-ligand
RUNX2
runt-related transcription factor 2
Smad
signaling mothers against decapentaplegic
TGFβ
transforming growth factor β
VEGF
vascular endothelial growth factor
11.1
Introduction
Bone formation and resorption are regulated by systemic and local factors acting in concert to maintain bone mass. Calciotropic and steroid hormones have been studied extensively for their effects on bone remodeling. However, there is compelling evidence to support the concept that systemic and locally produced growth factors play a central role in the regulation of bone remodeling. Growth factors regulate the replication, differentiation, and function of bone cells. This chapter will be limited to the description of factors that have a major effect on bone formation and osteoblastic function, whereas other chapters of this book will describe cytokines that regulate bone resorption. This is a somewhat arbitrary division since bone remodeling is coupled and osteoclastogenesis is dependent on osteoblastic signals. Furthermore, cytokines with primary effects on cells of the osteoclast lineage also play a role in the process of bone formation.
Skeletal cells express a variety of factors and regulatory signals. The skeletal growth factors most extensively studied for their effects in the skeleton include platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), transforming growth factor β (TGFβ), bone morphogenetic proteins (BMP), and insulin-like growth factors (IGFs). Some of these factors act as bone cell mitogens, and as such, they are important in the maintenance of an adequate number of skeletal cells. They also may increase bone cell replication when additional cells are needed, such as during fracture healing and repair. A factor may play a role in the differentiation of cells and osteoblastogenesis or may stimulate the differentiated function of mature cells. There are no known growth factors that are uniquely synthesized by skeletal cells, and those factors known as skeletal growth factors also are expressed in various nonskeletal tissues. However, growth factor regulation at the level of synthesis or activity may be targeted specifically in the skeleton by agents that act primarily on this tissue. Growth factors synthesized by skeletal cells may be present in the systemic circulation and act both as local and systemic regulators of bone remodeling. The source of circulating growth factors varies, although it is frequently the liver, the circulating platelets, or peripheral tissues. The systemic form of a factor can be regulated by agents and mechanisms different from those affecting the locally produced factor, and it is conceivable that the roles of the circulating and local form of a growth factor are not the same. This is not only because their synthesis is regulated by different hormones but also because they may become available under different physiological or pathophysiological circumstances. It is conceivable to believe that the local form of a growth factor plays a more immediate and possibly important role in the control of cell function since it has more direct access to its target cell. Locally synthesized growth factors can act either as autocrine factors and affect cells of the same class, or paracrine factors and affect different or adjacent cells.
In this chapter the function and regulation of selected growth factors will be discussed and their relevance to skeletal physiology will be considered.
11.2
Platelet-derived growth factor
PDGF was originally isolated from human platelets, and four members of the Pdgf gene family have been identified— Pdgfa , Pdgfb , Pdgfc , and Pdgfd . VEGF shares a high degree of sequence homology with PDGF, and these factors are often referred as members of the PDGF/VEGF family . This family of genes encodes a highly conserved cystine knot motif. Pdgfc and Pdgfd genes have a Clr/Cls, urchin endothelial growth factor, BMPl (CUB) domain linked to the cystine knot core motif by a hinge domain . Several reports have indicated that proteolytic release of the core from the CUB domain is required for the activation of PDGF C and PDGF D, although activity for the full-length peptide and independent activity for the CUB domain have been postulated . PDGFs must form homo or heterodimers to exhibit activity, and they can form PDGF AA, BB, AB, CC, or DD dimers . The various Pdgf genes are conspicuously expressed during development and in adult tissues. Transcripts for the Pdgfa , Pdgfb , and Pdgfc genes are detected in osteoblasts, but their basal expression is relatively low . PDGF D is synthesized by myocardial and vascular cells, where it induces cell proliferation and fibrosis, but there are no reports of PDGF D synthesis by the osteoblast . Since PDGF is present in circulating platelets, it can act like a local and systemic regulator of cell function . The five PDGF isoforms described can interact with either one of two PDGF receptors (PDGFRs), which have differential binding specificity for the various PDGF dimers . PDGFR α ligates PDGF A, B, and C chains and PDGFR β binds PDGF B and D chains . The two PDGFRs are structurally and functionally related, and PDGF binding results in receptor dimerization and the formation of PDGF αα, ββ, and αβ receptor dimers . For receptor activation, PDGF AA and PDGF CC require PDGFR αα or αβ dimers, PDGF DD requires PDGFR ββ or αβ dimers, whereas PDGF AB and PDGF BB can activate either PDGFR αα, αβ, or ββ dimers. PDGF also binds to matrix-associated and soluble molecules including α2-macroglobulin and PDGF-associated protein .
PDGF AA, AB, and BB are the isoforms studied most extensively in skeletal cells, and they exert similar biological actions. However, in skeletal as well as nonskeletal cells, PDGF BB is more potent than PDGF AA, and PDGF AB has intermediate activity . The primary action of PDGF in bone is the stimulation of cell replication. Histomorphometric analysis reveals an increase in cells of the osteoblastic lineage, but the effect is not specific and PDGF causes a generalized stimulation of cell replication in bone . The mitogenic effect is observed primarily in the periosteal layer, a zone rich in fibroblasts and preosteoblasts. It could be presumed that preosteoblastic cells, replicating under the influence of PDGF, eventually differentiate into mature osteoblasts. However, an inhibitory effect on the differentiation of stromal cells into cells of the osteoblastic lineage has been reported . This would indicate that the cells affected could remain in a proliferative undifferentiated state. Some cells, responding to other local signals, may differentiate, and as a consequence of the increased cell number, a modest increase in bone collagen synthesis is observed following exposure to PDGF. It is important to note that in accordance with the impaired cell differentiation, PDGF inhibits the expression of the mature osteoblastic phenotype, and decreases alkaline phosphatase activity and type I collagen mRNA levels . In calvariae, PDGF inhibits mineral apposition rates, confirming an inhibitory effect on osteoblastic function. These effects may be direct and indirect since PDGF inhibits the synthesis of IGF1 and IGF2 in osteoblasts, and IGF1 and IGF2 enhance osteoblastic function . PDGF enhances bone resorption by increasing the number of osteoclasts, an effect that may be secondary to an increase in the expression of Interleukin (IL) 6, a cytokine known to induce osteoclastogenesis . In agreement with its effects on bone resorption, PDGF increases the expression of matrix metalloproteinases (MMP) by the osteoblast . PDGF increases the rate of transcription of the Collagenase3 (MMP13) gene and stabilizes collagenase 3 transcripts in osteoblasts. MMP13 is a proteinase capable of initiating the degradation of type I collagen at neutral pH, and necessary to achieve a bone resorptive response to parathyroid hormone (PTH) .
Cells of the osteoblastic lineage express PDGF α and β receptors. PDGF binding to its osteoblast receptor results in receptor dimerization and activation of tyrosine kinase activity, leading to activation of protein kinase C and intracellular calcium signaling pathways . In rodent, but not in human osteoblasts, IL1 increases Pdgf α receptor transcripts and the binding and mitogenic activity of PDGF AA . TGFβ decreases PDGF binding . Information on the activity of PDGF CC and PDGF DD in skeletal cells is limited. PDGF CC interacts with PDGF αα and αβ receptor dimers and has a potent mitogenic activity for mesenchymal cells . PDGF CC induces the differentiation and regulation of endothelial cells and has potent angiogenic properties, directly or indirectly by upregulating VEGF . These properties of PDGF CC could be important during the vascularization of endochondral bone formation, as it has been described for VEGF. PDGF DD interacts with PDGF β receptors, has mitogenic activity for vascular cells, and induces tissue fibrosis, but its effects on the skeleton are not known .
Although there is considerable knowledge about the actions of PDGF in vitro, information about its effects on the skeleton in vivo are more limited. Consistent with some of its in vitro effects, the systemic administration of PDGF BB to ovariectomized rats prevents bone loss and increases the number of osteoblasts and bone formation . It is likely that the mitogenic effects of PDGF on preosteoblasts result in an increased number of osteoblasts, which are capable of forming bone. PDGF does not change osteoclast number when administered systemically to ovariectomized rats, although this may be related to this particular model, where the ovariectomy causes a substantial increase in bone resorption and remodeling, precluding an additional stimulatory effect by PDGF. Topical application of PDGF to craniotomy defects in rodents stimulates soft tissue repair but not osteogenesis . The effects of PDGF on endothelial cell proliferation and angiogenesis are likely beneficial to the process of wound healing, and PDGF accelerates the healing response of wounds due to an increase in cellularity and in the formation of granulation tissue .
Genetically engineered mice with gain or loss of function mutations have provided important information on the physiological role of PDGF during development and postnatally. Null mutations of Pdgfb , Pdgfa and b receptors cause embryonic lethality, and Pdgfa deletions cause prenatal and perinatal death . Therefore these models have not allowed for the study of the function of PDGF in the postnatal skeleton. Pdgfb and Pdgfb receptor null mice develop microvascular bleeding and absent vascular and mesangial cells in the renal glomerulus . Pdgfa and Pdgfa receptor null mutants have a defective alveolar formation in the lungs leading to emphysema and have reduced intestinal villi, thin dermis, and spermatogenic arrest, but Pdgfa null mutants do not manifest a skeletal phenotype . Pdgfc null mice exhibit neonatal lethality and numerous skeletal developmental abnormalities, including cleft palate and spina bifida . Similar defects are observed in Pdgfa receptor null mice, which exhibit a phenotype characterized by embryonic lethality, cleft face, spina bifida, and vascular and skeletal defects . Although PDGF B can interact with the PDGF α receptor, loss of function mutations of the Pdgfb gene do not resemble the Pdgfa receptor null phenotype, indicating that the functions of the PDGF α and β receptors are not redundant . The inactivation of the Pdgfd gene results in a mild vascular phenotype and the overexpression of PDGF DD, like that of PDGF CC, results in tissue fibrosis . This may be secondary to the mitogenic properties of PDGF CC and PDGF DD or due to the induction of tissue inhibitors of metalloproteinases.
The major source of PDGF is the systemic circulation, and skeletal cells probably become exposed to significant concentrations of PDGF following platelet aggregation. Nevertheless, skeletal cells express the Pdgfa , Pdgfb , and Pdgfc genes indicating that PDGF isoforms may act as autocrine regulators of skeletal cell function . PDGF BB secreted by preosteoclasts induces angiogenesis during bone remodeling and may promote osteogenesis and bone formation . The expression of the Pdgfa gene is enhanced by TGFβ and by PDGF, and that of Pdgfb enhanced by TGFβ . Following an initial induction of PDGF A, an autoregulatory mechanism may serve to maintain local levels of the growth factor. The regulation of Pdgf gene expression in skeletal cells is analogous to its regulation in nonskeletal cells indicating that there are no specific transcription factors responsible for the expression of PDGF in bone. Systemic hormones are not known to regulate Pdgf gene expression in osteoblasts. Since both TGFβ and PDGF are released by platelets following platelet aggregation, the subsequent induction of PDGF by these factors in the bone microenvironment may be a mechanism to ensure adequate levels of PDGF in skeletal tissues in conditions that follow platelet aggregation, such as fracture repair. Under basal conditions, there may be no need for skeletal cells to be exposed to significant concentrations of PDGF, and its levels are low .
11.3
Vascular endothelial growth factor
VEGF shares sequence homology and angiogenic properties with PDGFs, and often they are referred as members of the PDGF/VEGF family . The VEGF family includes VEGF A, VEGF B, VEGF C, VEGF D, VEGF E, and placenta growth factor . In the mouse, differential splicing results in various VEGF A isoforms. VEGF is expressed by a variety of cells. VEGF A is essential for angiogenesis and Vegfa and Vegf receptor 1 and 2 genes are expressed by chondrocytes and osteoblasts . VEGF receptor 1 acts as a signaling or a decoy receptor, and VEGF receptor 2 is the major signal-transducing receptor for VEGF . Both receptors seem to be positive regulators of skeletal development . VEGF A is required for blood vessel formation and vessel invasion into cartilage during the process of endochondral bone formation, and for chondrocyte survival during skeletal development . The expression of Vegfa by chondrocytes requires runt-related transcription factor 2 (Runx2), a transcription factor essential for normal osteogenesis . VEGF A is also required for intramembranous bone formation, it enhances osteoblastic maturation in vitro, and VEGF receptor 1 signaling is essential for osteoclast development . Vegfa null deletions are lethal due to defective hematopoiesis and defective blood vessel formation, but mice expressing one of three VEGF A isoforms, VEGF A 120, survive and their study has documented the function of VEGF A in endochondral bone formation and osteoblast maturation . VEGF also plays a critical role in craniofacial development. Bone histomorphometric analysis following adenoviral vector delivery of VEGF A also demonstrates a stimulatory effect of VEGF in osteoblast maturation, and in vivo VEGF A promotes angiogenesis and fracture repair . Osteoblast-derived VEGF plays a central role in bone formation during bone repair and optimal levels of VEGF are required for repair . VEGF enhances neo-vascularization as well as osteoclast formation and cartilage resorption during the repair process. VEGF also plays a suppressive role in adipogenesis and maintains adipocyte and osteoblast differentiation in balance, and a role in osteoclast differentiation and survival and bone resorption .
The expression of VEGF by osteoblasts is regulated by various growth factors. Activation of mitogen-activated protein (MAP) kinases by BMP, TGFβ, or FGF2 and activation of phosphatidylinositol 3 (PI3) kinase by IGF1 induce Vegf expression in osteoblasts . Following growth factor–induced synthesis, VEGF may serve to couple angiogenesis, and osteoblastic differentiation and function . The stimulatory effect of IGF1 on Vegfa gene expression is mediated by hypoxia-inducible factor (HIF). This may result in an increased vascular supply to the local environment and may serve as a protective mechanism in response to changes in oxygen availability to the osteoblast . It is of interest that the phenotypes of the conditional deletion of Hif1a and of Vegfa in cartilage are analogous . Hypoxia is a major driving signaling force for angiogenesis, and HIF1 is a ubiquitously expressed transcription factor that responds to hypoxia. HIF1 is the central regulator of adaptive responses to hypoxia and can induce the expression of a variety of target genes, including VEGF . Through this mechanism, HIF1 couples angiogenesis and plays a role in bone regeneration . HIF1a interacts with the transcription ATF4, and ATF4 is required for VEGF expression and angiogenesis . HIF1 also interacts with Notch, and hypoxia requires Notch signaling to maintain cells in the undifferentiated state . Notch plays a central role in osteoblastic cell fate and function and bone remodeling and may serve as an intermediary of hypoxia-mediated signals .
11.4
Fibroblast growth factor
FGFs form a family of ~18 structurally related secreted polypeptides, characterized by their affinity to glycosaminoglycan heparin-binding sites . FGFs bind to and activate four receptor tyrosine kinases. FGFs were initially isolated from the central nervous system and subsequently found in a variety of tissues, where they regulate growth and cell function, serving a role in tissue maintenance and repair . Skeletal cells synthesize both FGF1 and 2, the forms of FGF most extensively studied for their actions in the skeleton . Other forms of FGF, such as FGF8, have been reported to regulate the mitogenesis of cells of the osteoblastic lineage .
FGF has mitogenic activity in skeletal and nonskeletal cells and potent angiogenic properties . FGF increases a population of cells of the osteoblastic lineage, which differentiate into osteoblasts . However, FGF, like other potent mitogens, does not enhance the differentiated function of the osteoblast directly, and FGF2 inhibits alkaline phosphatase activity, type I collagen, osteocalcin, and osteopontin synthesis, independently of its stimulatory effects on osteoblastic cell growth . These effects are paralleled in vivo, and transgenic mice overexpressing FGF2 are osteopenic, although Fgf2 null mice exhibit impaired bone formation . The inhibitory effect of FGF2 on osteoblast differentiation is secondary to the inhibition of Wnt signaling, which is essential for osteoblastogenesis . FGF2 prevents the formation of the Wnt-dependent transcriptional complex formed by T-cell factor 4 and β-catenin. Confirming an inhibitory effect on osteoblastic function, FGF2 suppresses the synthesis of IGF1, a factor that stimulates the differentiated function of the osteoblast . FGF2 increases bone resorption by favoring osteoclastogenesis and stimulates the synthesis of MMP13 by the osteoblast . Marrow stromal cells from Fgf2 null mice exhibit decreased osteoclastogenesis in response to PTH. FGF2 induces TFGβ1 transcription, and TGFβ could mediate selected actions of FGF in bone .
The actions of FGF can be regulated by modifications in the affinity or number of FGF receptors (FGFR) in target cells. FGFR are a family of four distinct receptors, FGFR1–4 . FGFR are transmembrane protein responsive kinases with two to three immunoglobulin-like domains. Differential splicing of the extracellular region of FGFR can generate receptor variants with different ligand-binding specificity . The four known receptors, and variants, bind the members of the FGF family of polypeptides with different affinity and have different signaling and mitogenic potential. Activation of FGFR1, 2 and 3 by FGFs induce a mitogenic response, whereas activation of FGFR4 does not . Accordingly, mutations of the Fgfr1 , 2 , or 3 cause diverse skeletal syndromes including achondroplasia, a common cause of dwarfism . Studies on FGFR in preosteoblasts and osteoblasts have been limited. Activation of the signal transducers and activators of transcription 1 regulates FGFR in skeletal cells, suppresses FGFR3 expression in osteoblasts, and mediates FGF2 actions in chondrocytes . Cell surface and extracellular matrix heparan sulfate proteoglycans enhance the diffusion of FGFs and FGFR affinity to increase FGF signaling .
FGF is required for limb bud development, and the conditional inactivation of Fgfr1 and Fgfr2 in the limb bud in paired homeobox gene enhancer (Prx1) expressing cells results in severe skeletal hypoplasia . Similarly, the deletion of Fgfr1 and Fgfr2 in osteoprogenitor, Osterix expressing, cells results in impaired skeletal growth and osteopenia demonstrating that FGF plays a critical role in skeletal homeostasis . In vivo administration of FGF2 promotes bone and cartilage repair, and topical administration of FGF2 accelerates fracture healing in rodents and nonhuman primates . The mechanism is related to the mitogenic properties of FGF2 in skeletal cells in conjunction with its angiogenic activity. The observations that FGF2 is required for bone formation are contrasted by studies demonstrating that transgenic overexpression of FGF2 in the bone environment causes osteopenia . This may be related to an inhibitory effect of FGF2 on Wnt signaling, IGF1 expression, and osteoblastic function . Embryos harboring null mutations of the Fgfr2 die prior to skeletogenesis, but conditional inactivation of the Fgfr2 results in skeletal dwarfism secondary to decreased proliferation of osteoprogenitors without affecting osteoblast differentiation . These observations confirm early in vitro studies demonstrating a central role of FGF2 on preosteoblastic cell proliferation but not a stimulatory effect on osteoblastic function.
Investigations on the regulation of FGF synthesis in skeletal cells are limited to FGF2. In fibroblasts, heat shock induces the release of FGF1, suggesting that its secretion is related to cellular stress . The regulation of FGF2 expression in osteoblasts and nonskeletal fibroblasts is similar, and TGFβ1 and FGF2 increase FGF2 synthesis in osteoblasts . Sequence analysis of the FGF2 promoter reveals activator protein 1 (AP1) recognition sequences; therefore signals that induce Fos and Jun, components of the AP1 complex, have the potential to upregulate Fgf2 gene transcription . PDGF enhances the expression of FGF2 mRNA levels in fibroblasts and possibly in osteoblasts. Therefore skeletal growth factors with mitogenic properties, such as FGF2 and PDGF, are major inducers of FGF2 synthesis. PTH causes a transient induction of FGF2 transcripts in osteoblasts . FGF is stored in the extracellular matrix, where it is complexed by extracellular matrix and cell surface–associated heparan sulfate proteoglycans, which may prolong its half-life and allow future interactions with its receptors . Binding of FGF to heparan sulfate proteoglycans is a necessary step for the presentation of the factor to its receptor. The heparan sulfate proteoglycan syndecan is an integral membrane proteoglycan that binds FGF and components of the extracellular matrix, suggesting that it can regulate the effects of FGF2 on cell growth .
Mutations in FGFR1–3 are associated with skeletal dwarfism syndromes, craniosynostosis, and skeletal overgrowth syndromes . Achondroplasia, a common form of skeletal dwarfism, is associated with gain-of-function mutations in FGFR3 . Loss-of-function mutations in FGFR3 are associated with skeletal overgrowth and camptodactyly, tall stature, and hearing loss syndrome . Mutations in FGFR1 , FGFR2 , and FGFR3 resulting in FGF signal activation are associated with craniosynostosis in Apert, Crouzon, and related syndromes presenting with premature fusion of cranial sutures .
11.5
Transforming growth factor beta
TGFβ belongs to a family of closely related polypeptides with various degrees of structural homology and important effects on cell function . There are five Tgfb genes, and mammalian cells express genes encoding TGFβ1, 2, and 3. Bone matrix contains TGFβ1, 2, and 3 homodimers as well as 1.2 and 2.3 heterodimers . TGFβ1, 2, and 3 have similar effects on bone cell function although their potency differs . TGFβ stimulates DNA synthesis and cell replication and has a modest stimulatory effect on collagen synthesis in calvariae . Bone histomorphometric analysis of intact rat calvariae exposed to TGFβ demonstrates a stimulatory effect on mineral apposition rates, and in vivo studies have confirmed a stimulatory effect of TGFβ on bone formation . The effects of TGFβ on cell replication and the differentiated function of the osteoblast are dependent on the target cell, its state of differentiation, and the culture conditions used. In primary cultures of rat osteoblasts, TGFβ has a biphasic stimulatory effect on DNA synthesis, whereas in rat osteosarcoma cells, it inhibits cell growth . In osteoblasts, TGFβ inhibits alkaline phosphatase activity and osteocalcin synthesis suggesting an inhibitory effect on the differentiated function of the osteoblast. On the other hand, TGFβ stimulates collagen synthesis by increasing type 1 collagen col1a1 gene transcription . In osteosarcoma cells, TGFβ increases type I collagen, fibronectin, osteonectin, and osteopontin mRNA expression . In stromal cell cultures, TGFβ favors chondrocytic differentiation and opposes the effect of BMP on osteoblastogenesis . Consequently, whereas TGFβ stimulates selected parameters of osteoblastic function, it does not direct the maturation of undifferentiated cells toward mature osteoblasts and seems more relevant to chondrogenesis than to osteoblastogenesis. The actions of TGFβ on bone resorption have been a source of controversy. TGFβ has a biphasic effect on osteoclastogenesis. At low concentrations, it enhances osteoclast formation, whereas at high concentrations, it is inhibitory. The stimulatory effect on osteoclast formation seems to be related to the induction of prostaglandins, and the inhibitory effect secondary to a decrease in the differentiation of cells of the osteoclast lineage with a consequent decrease in bone resorption . However, TGFβ plays a key role in the development of osteolytic metastases since it stimulates tumor cells to release cytokines that resorb bone adjacent to tumor cells . Recently, TGFβ was found to induce the expression of the Notch ligand Jagged1 in carcinoma of the breast cells, and as a consequence to enhance Notch signaling and bone resorption following the induction of IL6 by Notch . Osteoblasts express type I and II TGFβ receptors, and following TGFβ–surface-receptor interactions, TGFβ signals through the signaling of mothers against decapentaplegic (Smad) pathway, resulting in the phosphorylation of Smad2 and 3, their association with Smad4, and nuclear translocation to regulate transcription . PTH and glucocorticoids modify TGFβ binding to its receptors on osteoblasts . Glucocorticoids shift the binding of TGFβ from type I and II receptors to betaglycan, which is not a signal-transducing molecule .
In accordance with its actions in vitro the systemic administration of TGFβ2 to experimental animals stimulates cancellous bone formation, and subperiosteal injections of TGFβ1 and 2 induce osteogenesis and chondrogenesis in rat femurs . In contrast, transgenic mice overexpressing TGFβ2 under the control of the osteoblast-specific bone gamma-carboxyglutamate protein ( Bglap ) promoter exhibit osteopenia . Although TGFβ induces chondrocyte differentiation, suppression of TGFβ signaling in vivo also favors terminal chondrocyte differentiation suggesting a dual role of TGFβ in chondrocyte maturation . Targeted gene disruption of the Tgfb1 gene in mice does not result in changes in skeletal development . This may be secondary to an early lethal phenotype of Tgfb1 null mice, due to a severe inflammatory disease. Conditional deletion of the Tgfb type2 receptor in chondrogenic cells results in skeletal developmental defects . TGFβ enhances soft tissue wound healing, and like FGF2, it may play a role in fracture repair . However, its cytostatic actions and its fibrosis-inducing properties would limit its topical and systemic use in the treatment of metabolic bone disorders .
TGFβ is secreted as a latent high molecular weight complex consisting of the carboxy-terminal remnant of the TGFβ precursor and a TGFβ binding protein . The biologically active levels of TGFβ depend on changes in its synthesis and in the activation of its latent form. By inducing lysosomal proteases, bone regulatory agents increase the levels of biologically active TGFβ in bone . FGF, TGFβ itself, and estrogens increase TGFβ1 synthesis in osteoblasts, and ovariectomy reduces the concentration of TGFβ in rodent bone . The latter may suggest a role of TGFβ in the abnormalities observed in the estrogen-deficient state. There have been limited studies on the regulation of TGFβ2 and TGFβ3 synthesis in osteoblasts, but the Tgfb2 and Tgfb3 gene promoters contain cyclic adenosine monophosphate (AMP) responsive elements, indicating a potential regulation by cyclic AMP inducers . The bioavailability of TGFβ is controlled by the extracellular matrix proteins fibrillin 1 and 2. By controlling the local bioavailability of TGFβ and BMP, fibrillins maintain osteoblast function and bone formation . Fibrillins also control the TGFβ-dependent induction of receptor activator of nuclear factor–κ-B-ligand (RANKL) by osteoblasts and bone resorption . It is of interest that mutations in genes encoding fibrillin 1 and 2 are associated with Marfan’s syndrome and the related congenital contractural arachnodactyly .
11.6
Bone morphogenetic proteins
BMPs are members of the TGFβ superfamily of polypeptides and were originally identified because of their ability to induce endochondral bone formation . BMPs account for most of the TGFβ superfamily of peptides, and the proteins display extensive conservation among species . BMP1 is a protease, which cleaves procollagen fibrils and is unrelated to other BMPs . BMP3, or osteogenin, is different from other BMPs since it lacks their osteogenetic properties, and it inhibits osteogenesis and opposes BMP2 actions . Although BMPs are synthesized by skeletal cells, their synthesis is not limited to bone, and they are expressed by a variety of extraskeletal tissues, where they play a critical role in developmental and cellular functions. BMP1–6 are expressed by osteoblasts, but the degree of expression depends on the cell line examined and its stage of differentiation . BMP2, 4, and 6 are the most readily detectable BMPs in osteoblasts, and BMP2 and 4 are 92% identical in their amino acid sequence and have virtually identical biological activities. Experiments using kinase-deficient truncated BMP receptors or using BMP antagonists have demonstrated that the locally synthesized BMPs play an autocrine role in osteoblast cell differentiation and function .
A function of BMPs is the induction of mesenchymal cell differentiation toward cells of the osteoblast lineage and the promotion of osteoblastic activity . As osteoblasts undergo terminal differentiation, they undergo apoptosis, an expected result of cell maturation . Consequently, BMPs favor osteoblastic cell death. MicroRNAs play a role in the posttranscriptional control of gene expression by catalyzing the cleavage of messenger (m)RNAs or by repressing mRNA translation . MicroRNAs have emerged as key regulators of diverse biological and pathological processes, and they play a central role in the actions of BMPs on bone cell determination . The genesis and differentiation of bone-forming osteoblasts and bone-resorbing osteoclasts are coordinated events. RANKL is synthesized by osteoblasts and osteocytes, and in conjunction with colony-stimulating factor 1, it induces osteoclast maturation . Osteoprotegerin, a secreted receptor of the tumor necrosis factor receptor family, acts as a decoy receptor that binds RANKL, precluding RANKL binding to RANK and its effects on osteoclastogenesis and bone resorption. BMPs stimulate bone resorption and play a direct and indirect role in osteoclastogenesis. By inducing osteoblast maturation, there is increased RANKL availability. In addition, BMPs sensitize osteoclasts to the effects of RANKL on osteoclastogenesis and osteoclast survival . Consequently, overexpression of BMP4 in osteoblasts causes osteopenia and increased osteoclast number . Conversely, inactivation of BMP signaling in osteoblasts suppresses osteoclastogenesis and surprisingly induces osteoblastogenesis, questioning the stimulatory role of BMPs in osteoblast differentiation and bone formation . BMP signaling induces the synthesis of the Wnt signaling antagonists sclerostin and Dickkopf1 explaining the impaired osteoblastic differentiation . The decrease in Wnt signaling by BMPs contributes to their stimulatory effect on bone resorption since Wnt signaling suppresses osteoclastogenesis . BMPs also induce osteoprotegerin transcription, and this may be a protective mechanism to temper their effects on osteoclastogenesis . BMPs inhibit MMP13 expression in osteoblasts, a matrix metalloprotease required for normal bone resorption .
BMPs induce endochondral ossification and chondrogenesis . BMPs stimulate chondrocyte maturation and function, increasing the expression of type II and type X collagens and the incorporation of sulfate into glycosaminoglycans . Overexpression of BMP2 and 4 in developing limbs results in an increase in chondrocyte number and in matrix cartilage, which may lead to joint fusions . The anabolic effects of Indian and Sonic hedgehog and BMP2 and 4 in metatarsal cultures are analogous, and BMPs mediate their actions on endochondral ossification . Whereas BMPs induce chondrogenesis, they prevent terminal differentiation of myogenic cells, inhibiting the transcription of the muscle-specific nuclear factors MyoD and myogenin . BMPs act in conjunction with other growth factors and increase the pool of IGF1 producing and IGF1 target cells .
BMPs signal by interacting with type I and type II receptors. BMPs can signal by interacting with four type I receptors, namely, activin receptor-like kinase (ALK)1/activin A receptor (ACVR)L1, ALK2/ACVR1, ALK3/BMPRIA, and ALK6/BMPRIB, and three type II receptors, namely, BMPRII, ActRII/ACVR2, and ActRIIB/ACVR2B .
Mutations of the type 1 BMP receptor ACVR1 or ALK2 that enable activation to trigger signaling cause fibrodysplasia ossificans progressiva . Upon ligand binding and activation of the type I receptor, dimers of the type I and type II receptor initiate a signal transduction cascade, activating the Smad or the MAP kinase signaling pathways . Following receptor activation by BMPs, Smad1, 5, and 8 are phosphorylated at serine residues and translocated into the nucleus following heterodimerization with Smad4 . In the nucleus, Smads can bind to DNA sequences directly, bind and cooperate with other transcription factors, or bind and displace nuclear factors from their DNA binding sites. MAP kinase signaling results in P38 MAP kinase or extracellular regulated kinase (ERK) activation by BMPs . In cells of the osteoblastic lineage, the Smad signaling pathway appears to prevail over MAP kinase signaling .
The transcriptional and posttranscriptional regulation of BMP expression in osteoblasts reveals autoregulation of BMP synthesis since BMP2 and 4 mRNA levels are BMP dependent . BMPs cause an early, short-lived, induction of BMP4 mRNA in osteoblasts followed by an inhibitory effect. A positive feedback loop regulating BMP2 and 4 expressions involving Runx2 is possible since BMPs induce Runx2 expression, and the BMP2 and 4 promoters contain Runx2 binding sequences. BMP6 expression in osteoblasts is steroid dependent, and BMP6 mRNA levels are induced by estrogens .
Mice deficient in Bmp2 are not viable due to placental and developmental defects, and the Bmp4 null mutation is lethal between 6.5 and 9.5 days of gestation due to a lack of mesodermal differentiation and patterning defects . Mice with disruptions of the BMP signaling Smad5 gene also develop multiple embryonic defects, some reminiscent of those observed in Bmp2 null mutants . The lethality of these mutations has prevented the assessment of the impact of BMP2 and 4 on the adult skeleton. The Bmp6 null mutation is not lethal, and skeletogenesis is normal except for delayed ossification in the sternum . Bmp gene inactivation results in significant developmental defects outside the skeleton; Bmp7 null mice display a lack of eye and glomerular development and Bmp8 null mutations result in defective spermatogenesis .
BMP activity is regulated by a large group of secreted polypeptides that bind and limit BMP action ( Table 11.1 ). These extracellular BMP antagonists prevent BMP signaling by binding BMPs and precluding their interaction with cell surface receptors . Extracellular BMP antagonists are classified into three families: (1) Noggin and chordin families; (2) twisted gastrulation (Tsg); and (3) Dan/Cerberus family . The tissue expression pattern of BMP antagonists is specific to each antagonist. Skeletal cells express noggin, chordin, chordin-like, follistatin and related gene, Tsg, gremlin 1 and gremlin 2 or the protein related to Dan and Cerberus. The primary function of BMP antagonists is to control and temper BMP activity, although the BMPs targeted by each antagonist are not uniformly the same. Ectodin is expressed in tooth enamel . In addition, selected BMP antagonists, such as sclerostin and Coco, also target Wnt signaling receptors/coreceptors . Sclerostin can bind selected BMPs, but it is primarily a Wnt antagonist that interacts with Wnt coreceptors. Sclerostin is predominantly expressed in osteocytes, where it is downregulated by PTH . Overexpression of noggin and gremlin1, two classic BMP antagonists, or sclerostin, in the skeletal microenvironment prevents osteoblastic differentiation in vitro, and in vivo causes osteopenia secondary to decreased bone formation . The global inactivation of Nog causes intrauterine lethality and articular developmental defects leading to joint fusions . In vitro, Nog inactivation facilitates osteogenesis . However, the conditional inactivation of Nog in the osteoblast lineage causes osteopenia secondary to increased bone resorption . These observations confirm the stimulatory effect of BMPs in bone resorption and suggest that BMPs in excess may have a detrimental effect on bone homeostasis . The global inactivation of Grem11 causes osteopenia, possibly due to skeletal developmental defects, but its conditional inactivation in osteoblasts causes a transient increase in bone formation and bone mass . Tsg has BMP agonist and antagonist activities, and it is required for normal skeletal development but not for postnatal skeletal homeostasis . Overexpression of Tsg inhibits BMP activity in cells of the osteoblast lineage, preventing their differentiation and cell maturation . It is of interest that the synthesis of many BMP antagonists, such as thatof noggin and gremlin, is induced by BMPs in osteoblasts, suggesting the existence of a protective mechanism to prevent skeletal cells from excessive exposure to BMPs .
|
Members of the Cyr61, connective tissue growth factor (CTGF) and Nephroblastoma overexpressed (Nov) (CCN) family, are structurally related to certain BMP antagonists, such as Tsg and chordin . CCN proteins are highly conserved and share four distinct structural modules: an IGF-binding domain, a von Willebrand type C domain containing the cysteine-rich domain, a thrombospondin-1 domain, and a carboxyl-terminal domain . CTGF binds to BMP2 and 4 through its cysteine-rich domain and binds to Wnt coreceptors through its carboxyl-terminal domain, and its overexpression modifies BMP and Wnt signaling and effects on osteoblastogenesis . Transgenic overexpression of CTGF in osteoblasts decreases bone formation and causes osteopenia . Similarly, Nov binds BMP2 and 4, and its overexpression inhibits osteoblastogenesis and causes osteopenia . The structural similarities with BMP antagonists of the Tsg and chordin families, and these observations indicate a potential functional relationship between CCN peptides and more classic extracellular BMP antagonists. The global inactivation of Nov in mice does not cause a skeletal phenotype, and the global inactivation of Ctgf leads to defective endochondral bone formation and perinatal lethality . The conditional inactivation of Ctgf in the limb bud confirms a role in endochondral bone formation, and its conditional inactivation in osteoblasts suggests a modest role of CTGF in skeletal homeostasis . Cyr61 is a target of Wnt signaling, induces BMP2 expression, and promotes osteogenic differentiation and mesenchymal cell migration . The conditional deletion of Cyr61 in the osteoblast lineage causes osteopenia secondary to decreased osteoblast number and function associated with increased RANKL expression and osteoclast number . These effects were associated with decreased VEGFA and vasculature, increased sclerostin, and decreased Wnt signaling suggesting that Cyr61 is a modulator of Wnt activity.
11.7
Insulin-like growth factor
IGF1 and IGF2 have structural similarities with proinsulin and are considered essential for normal cell growth and function in multiple tissues including bone . IGF1 and IGF2 are present in the systemic circulation and are secreted by the osteoblast. In the circulation, IGF1 forms a large molecular weight complex with IGF binding proteins (IGFBPs) and the acid-labile subunit . Although several IGFBPs form the complex, IGFBP3 is the most abundant, and systemic IGF1 and IGFBP3 levels are growth hormone (GH) dependent. IGFBPs are present in the circulation at concentrations in excess of those of IGF1. Consequently, there is little free IGF1 in plasma. Systemic IGF1 is mostly synthesized in the liver, and it is responsible for the growth-promoting effects of GH in most tissues . It is important to note that IGF1 is synthesized by multiple peripheral tissues, where it is regulated by other hormones, and only to a minor extent by GH. In addition to their function as systemic regulators of growth, IGF1 and IGF2 play an important role in the autocrine and paracrine regulation of cell metabolism in a variety of tissues, including bone . IGF1 and IGF2 are the most prevalent growth factors present in the skeletal tissue.
IGF1 and IGF2 have similar effects on bone formation, although IGF1 is more potent than IGF2 . IGFs are modest mitogens, increasing the replication of preosteoblastic cells, which presumably differentiate into mature osteoblasts. The most important function of IGFs is to enhance the differentiated function of the osteoblast. IGFs stimulate type I collagen transcription, an effect independent from their mitogenic actions, and increase mineral apposition rates . IGF1 and IGF2 inhibit MMP13 synthesis by the osteoblast and as a consequence decrease bone collagen degradation . IGF1 and IGF2 are important in the maintenance of the differentiated osteoblast phenotype. IGFs not only enhance osteoblastic function, but their synthesis is also differentially regulated by factors that stimulate or inhibit the differentiated expression of the osteoblastic phenotype . A similar role has been suggested for IGF2 in myoblasts, where its expression follows that of genes that are determinants of myogenic differentiation . It is important to note that whereas IGF1 stimulates the differentiated function of the osteoblast, its role in the differentiation of undifferentiated mesenchymal cells toward the osteoblastic lineage is controversial . Indirectly, IGF1 can favor osteoblastogenesis by stabilizing β-catenin consequently enhancing Wnt canonical signaling, which is essential for osteoblastogenesis . Although the primary role of IGFs is the stimulation of bone formation, IGF1 can increase the synthesis of RANKL by osteoblasts and, in this way, enhance osteoclast recruitment, although in vivo experiments have demonstrated an inconsistent impact of IGF1 on osteoclastogenesis . Igf1 null mice exhibit a decreased number of functional osteoclasts indicating that IGF1 is required for normal osteoclastogenesis .
Skeletal cells express the IGF1 and IGF2 receptors, and the IGF1 receptor mediates their anabolic actions. This receptor is a transmembrane glycoprotein tetramer with ligand-activated tyrosine kinase activity. Insulin receptor substrate (IRS) 1 and 2 are well-characterized substrates for the IGF receptor tyrosine kinase, mediating the effects of IGF1 . In osteoblasts, tyrosine phosphorylation of IRS molecules by the activated receptor results in the activation of PI3 kinase-phospho Akt signaling or in the activation of the MAP kinases, p38, Jun-N-terminal kinases and ERK1/2 . The IGF2 receptor is the same as the mannose-6-phosphate receptor, does not have a function in IGF signal transduction and clears IGF2, regulating its levels during fetal development . IGF1 receptor number in osteoblasts can be modulated by various agents known to regulate bone cell metabolism, including PDGF, glucocorticoids, and 1,25-dihydroxyvitamin D 3 .
IGF1 has been tested for its effects on bone metabolism in vivo in experimental animals and humans. IGF1 increases bone formation and prevents trabecular bone loss in experimental conditions of skeletal unloading and increases bone mineral density (BMD) in ovariectomized animals . The short-term administration of IGF1 to normal humans results in an increase in serum levels of type I procollagen peptide and an increase in the excretion of collagen crosslinks, demonstrating an increase in bone turnover . IGF1 increases BMD in patients with osteopenia secondary to anorexia nervosa, but the potential use of IGF1 in humans may be limited by possible side effects and the lack of skeletal specificity . The content of IGF1 in human cortical bone decreases with age, a decline that parallels the one observed in serum concentrations of IGF1 with aging . Consequently, it is not known whether this is due to a decrease in skeletal IGF1 accumulation from the systemic circulation, or due to a decrease in the synthesis of IGF1 by the aging skeleton.
The study of genetically engineered mice has provided additional insight into the action of IGF1 in vivo . Transgenic mice expressing IGF1 in osteoblasts under the control of the Bglap promoter exhibit a transient increase in trabecular bone volume secondary to an increase in osteoblast function and bone formation . In addition, mice expressing IGF1 under the control of the Col1a1 promoter exhibit increased bone remodeling . Igf1 and Igf1 receptor global gene deletions have provided valuable information on the role of IGF1 during development, and conditional gene deletions have provided information on the effects of IGF1 in the adult skeleton . Igf1 deletion causes a reduction in chondrocyte maturation, and femoral length, and osteopenia secondary to a decrease in bone formation . Mice carrying mutations of the Gh-releasing hormone receptor or of the Ghr have absent GH secretion or action, and consequently low serum IGF1 . These models allow for the determination of the contribution of systemic IGF1 to the skeleton, and the phenotype of either mutant is characterized by small growth plates, osteopenia, and reduced cortical bone, but normal trabecular bone volume. This suggests a more pronounced role of systemic IGF1 on cortical than on trabecular bone. However, mice harboring a liver-specific deletion of the Ghr exhibit marked decreases in serum IGF1 levels and cancellous bone osteopenia . Mice carrying a liver-specific Igf1 deletion display a 75% reduction in total serum IGF1 levels, normal free IGF1 levels, and absent skeletal phenotype attributed to the normal serum levels of free IGF1 and the extra hepatic synthesis of IGF1 . However, mice carrying dual deletions of Igf1 and the Als display marked reduction in total serum IGF1 and a significant reduction in cortical bone volume and bone strength . Igf1r null mice die after birth and demonstrate severe growth retardation, and the conditional disruption of the Igf1r gene selectively in osteoblastic cells causes a decrease in osteoblast number and impaired bone formation resulting in reduced trabecular bone volume . This observation documents the fundamental role played by IGF1 in the maintenance of bone formation and structure. Accordingly, deletion of the Irs1 or 2 gene causes osteopenia . However, the phenotypes are not identical and Irs1 null mice exhibit low bone turnover osteopenia and do not respond to PTH, whereas Igf2 null mice exhibit increased bone resorption and respond to the anabolic effects of PTH in bone . It is of interest that a spontaneous mutation of Irs1 in mice results in hyperinsulinemia, reduced growth, and osteopenia due to impaired IGF1 signaling and decreased bone formation . The various models described confirm the anabolic function of IGF1 in bone. Skeletal derived IGF1 might play a more important role in the maintenance of trabecular bone, and systemic IGF1 might be more important in the regulation of cortical bone. The preferential deletion of Igf1 or Igf1r in osteocytes, achieved by crossing conditional Igf1 or Igf1r mice with transgenics expressing Cre under the control of the dentin matrix protein ( Dmp ) 1 promoter, results in cortical osteopenia .
The Igf1 gene is complex, contains six exons, and has alternate promoters in exons 1 and 2 . The exon 1 promoter has four transcription initiation sites and is responsible for the regulation of IGF1 expression in most extrahepatic tissues including bone . The IGF1 exon 2 promoter has two transcription initiation sites and is responsible for the transcriptional regulation of IGF1 by GH in the liver . IGF1 exon 2 is minimally expressed by osteoblasts, and GH is not a major inducer of IGF1 in these cells . Hormones and growth factors regulate the synthesis of IGF1 in osteoblasts, and PTH, PTH-related peptide, and other inducers of cyclic AMP in osteoblasts increase IGF1 synthesis . IGF1 mediates selected anabolic actions of PTH in bone in vitro and in vivo . The stimulatory effect of PTH on collagen synthesis in vitro is decreased by IGF1 neutralizing antibodies, and the stimulatory effect of PTH on bone formation in vivo is not observed in Igf1 or Irs1 null mice . These observations do not exclude the possibility that other factors mediate selected actions of PTH on the skeleton. For example, PTH can suppress the activation of Notch signaling that is an inhibitor of osteoblast differentiation . PTH also increases the levels of, and activates, skeletal TGFβ, which could mediate selected effects of PTH in bone . Estrogens increase and glucocorticoids inhibit IGF1 synthesis in osteoblasts . Selected inhibitory effects of glucocorticoids on bone metabolism can be explained by reduced IGF1 levels in the bone microenvironment. However, glucocorticoids also inhibit osteoblast gene expression and function directly. In addition to hormones, skeletal growth factors regulate IGF1 synthesis. PDGF, FGF2, and TGFβ1 decrease IGF1 transcripts and polypeptide levels in osteoblasts, and this inhibition of IGF1 synthesis correlates with their inhibitory actions on osteoblastic differentiated function . In contrast, BMP2, an agent that enhances osteoblast differentiation and function, increases IGF1 synthesis in osteoblasts . IGF1 expression also is controlled by posttranscriptional mechanisms. Nocturnin, a circadian gene with peak expression at light offset, interacts with IGF1 and decreases IGF1 expression by posttranscriptional mechanisms so that IGF1 expression in the skeleton is lowest at night .
The Igf2 gene is complex and contains four promoters, and in osteoblasts, like in hepatocytes, IGF2 expression is under the control of the IGF2 P3 promoter . Hormones do not modify IGF2 synthesis in skeletal cells. The skeletal growth factors, FGF2, PDGF, and TGFβ1, inhibit IGF2 transcription by inhibiting IGF2 P3 activity, an effect analogous to their inhibition of IGF1 expression, demonstrating a coordinated suppression of IGF1 and IGF2 synthesis by mitogenic growth factors .
11.8
Insulin-like growth factor binding proteins
IGFBPs are a family of related proteins known to bind specifically IGF1 and IGF2. There are six classic IGFBPs, termed IGFBP1–6 . In addition, members of the CCN family of proteins have an IGFBP domain and can bind IGFs . There are significant structural similarities in IGFBPs among species, indicating a high degree of evolutionary conservation. Osteoblasts express IGFBP1–6 transcripts . IGFBPs regulate the bioavailability of IGFs and prevent their degradation. IGFBPs can potentiate or inhibit the effects of IGF1 and IGF2 on cell function and vary in their affinity for IGF1 and IGF2 . The binding of IGFs to IGFBPs may sequester the growth factor and preclude its interactions with cell surface receptors, although IGFBPs associated with the extracellular matrix may increase the local effective concentration of the growth factor and potentiate its effects . In addition, IGFBPs may have IGF-independent effects and regulate cellular events directly.
IGFBP2 is important in the storage and transport of IGFs, and IGFBP2 serum levels correlate with BMD in humans . In vitro, IGFBP2 prevents the effects of IGF1 on osteoblast function, and overexpression of IGFBP2 under the control of the cytomegalovirus (CMV) promoter leads to small mice with decreased bone mineral content and failure to respond to the anabolic effects of GH in bone . Accordingly, the global inactivation of Igfbp2 in female mice causes an increase in cortical bone . However, male Igfbp2 null mice exhibit reduced cortical and trabecular bone, impaired osteoblastic differentiation, and bone formation indicating that IGFBP2 is necessary for bone remodeling in male mice. The mechanism involves the suppression of phosphatase and tensin homolog and the induction of pАkt and as a consequence stabilization of β-catenin and increased Wnt signaling . IGFBP3 is the major component of the IGF complex in serum and, like the circulating IGF1, is GH dependent . IGFBP3 can inhibit or stimulate IGF activities, the latter by upregulating IGF1 delivery to cell surface receptors. Overexpression of IGFBP3 under the control of the CMV or phosphoglycerate kinase promoter causes growth retardation and osteopenia . Ablation of Igfbp3 results in osteopenia associated with increased RANKL . IGBP4 is an IGF1 inhibitory binding protein, but under specific conditions, IGFBP4 and IGFBP5 were reported to stimulate bone cell function independent from their interactions with IGF1 . However, transgenic mice overexpressing either IGFBP4 or IGFBP5 under the control of the osteoblast-specific Bglap promoter exhibit osteopenia and decreased bone formation . The osteopenia is probably secondary to sequestration of the IGF1 present in the bone microenvironment inhibiting its biological activity. It is possible that the different effects reported for IGFBP4 and 5 are dependent on their interactions with extracellular matrix proteins or on the levels of IGFBP present in a specific tissue. However, both transgenic models indicate that, when in excess, IGFBP4 and IGFBP5 are inhibitory proteins in the skeletal environment. This was documented using retroviral vectors to overexpress IGFBP5 in osteoblasts, which provoked an inhibition of osteoblastic cell function . IGFBP1 is important for glucose homeostasis, and there is limited information on the function of IGFBP6 in skeletal tissue.
Although the six IGFBPs are expressed by skeletal and nonskeletal cells, their basal and regulated expression is cell specific . In vitro studies indicate that the pattern of IGFBP expression is dependent on the stages of osteoblast differentiation. IGFBP2 and 5 expressions are highest in the proliferative phase of rat osteoblastic cell cultures, and IGFBP3, 4, and 6 expression peaks during the maturation phase . The regulation of IGFBP expression during osteoblastic cell differentiation may be related to the relative levels of autocrine and paracrine factors present in the cellular environment. IGFs increase osteoblast IGFBP5 expression, whereas growth factors with mitogenic activity inhibit IGFBP5 and stimulate IGFBP4 expression .
In addition to local autocrine and paracrine factors, systemic hormones modulate IGFBP synthesis; however, the effects appear to be cell line and culture condition dependent . GH increases IGFBP3 in normal rat osteoblasts but not in osteosarcoma cells, and cyclic AMP inducers increase the synthesis of IGFBP3, 4, and 5 . Since agents that induce cyclic AMP in osteoblasts also stimulate the synthesis of IGF1, the induction of the binding proteins may be a mechanism to control overexposure of cells to the newly synthesized IGF1. Conversely, glucocorticoids inhibit the synthesis of IGF1, IGFBP3, 4, and 5, although they increase the expression of IGFBP2 in osteoblasts . 1,25-Dihydroxyvitamin D 3 increases osteoblast IGFBP3 and 4 expression .
The abundance of IGFBPs in the extracellular space can be regulated by proteolytic degradation. IGFBP proteases have been characterized by diverse sources, including osteoblasts, which secrete MMPs and serine proteases . Pregnancy-associated plasma protein A (PAPPA) is a metalloproteinase expressed by skeletal cells that play a role in osteoblast function by modulating IGF1 bioavailability . IGFBP2 and 5 also are substrates for PAPPA. Addition of PAPPA to osteoblast cultures promotes cell proliferation by increasing IGF1 bioavailability, and transgenic mice expressing PAPPA under the control of the Col1a1 promoter exhibit increased bone formation . Pappa null mice lack IGFBP4 proteolytic activity, and at birth, they exhibit dwarfism secondary to the sequestration of IGF by IGFBP4 . Postnatally, Pappa null mice exhibit decreased trabecular bone volume, low bone turnover osteopenia, and delayed fracture healing . These observations are consistent with a decrease in bioavailable IGF1 in the bone environment due to excessive IGFBP4. The expression of PAPPA in osteoblasts is enhanced by TGFβ, agents that induce cAMP, such as PTH, and cytokines, such as IL1 and 4 and TNFα . The synthesis and activity of other metalloproteinases, such as MMP13, in osteoblasts also are regulated by cytokines and hormones, including IL6 and glucocorticoids . Limited proteolysis of IGFBPs by serine proteases controls the bioavailability of IGF1 in the circulation by cleaving IGFBP3. In addition, IGFBP3 is cleaved by plasmin, and the bioavailability of IGF1 in the bone microenvironment can be regulated by activators and inhibitors of plasminogen .
11.9
Hepatocyte growth factor
Hepatocyte growth factor (HGF) also known as Scatter factor is a large molecular weight polypeptide known for its angiogenic and mitogenic properties . HGF plays a role in liver and kidney repair and in the development of bone metastases . HGF signals via the product of the proto-oncogene c-met, a tyrosine kinase-activated receptor, and HGF and c-met are expressed by mesenchymal cells, osteoblasts, and osteoclasts . HGF is mitogenic for cells of the osteoblast and osteoclast lineages, and its synthesis by the osteoblast is enhanced by growth factors with a role in wound healing and fracture repair . Therefore HGF may play a role in bone remodeling and repair. Studies in transgenic mice expressing HGF under the control of the metallothionein 1 ( Mt1 ) promoter and studies in Hgf or Cmet null mice have revealed that HGF is required for muscle cell migration . Hgf null mutants fail to form normal muscles. Due to embryonic lethality, the skeletal phenotype was not examined. FGF2 and PDGF increase the synthesis of HGF by osteoblasts, whereas glucocorticoids are inhibitory . Since HGF plays a role in mitogenesis and tissue repair, inhibition of its synthesis by glucocorticoids may be relevant to the inhibitory effect of these steroids on wound healing and tissue repair. HGF and c-met are expressed at the site of fractured bone, and HGF induces BMP receptors . However, HGF can reduce BMP signaling and activity in vivo and in vitro .
Acknowledgments
The author of this chapter is currently supported by grants from the National Institutes of Health DK045227, AR063049, AR068160, AR072987 and AR076747. The author thanks Ms. Mary Yurczak for valuable secretarial assistance.
References
Related posts:
 Osteoclast biology
Osteoclast biology
 Impact of physical characteristics and lifestyle factors on bone density and fractures
Impact of physical characteristics and lifestyle factors on bone density and fractures
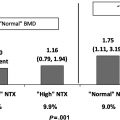 Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
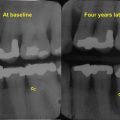 Relationship between periodontal disease, tooth loss, and osteoporosis
Relationship between periodontal disease, tooth loss, and osteoporosis
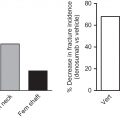 On the evolution and contemporary roles of bone remodeling
On the evolution and contemporary roles of bone remodeling
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


