Skeletal Dysplasias
Danielle A. Katz
Polyostotic Fibrous Dysplasia and Albright’s Syndrome
Fibrous dysplasia is a condition in which there is fibro-osseous tissue in place of normal lamellar bone. Most often (85%) this process is monostotic (affecting a single bone), but it may be polyostotic (involving multiple bones). Cutaneous markings and endocrinopathies may accompany the polyostotic form, which usually has more severe skeletal involvement. This section will focus on the polyostotic form.
Pathogenesis
Etiology
Not inherited
May be due to failure of woven bone to mature to lamellar bone
G-protein gene mutations in both monostotic and polyostotic fibrous dysplasia, Albright’s syndrome, and solitary pituitary adenoma
Activating (gain of function) mutations in GNAS1 (encodes alpha subunit of stimulatory G protein)
Epidemiology
Female predominance
Diagnosis usually made in late childhood or early adolescence
30% to 50% of patients with polyostotic fibrous dysplasia have Albright’s syndrome.
Pathophysiology
Fibro-osseous tissue within bone, most often metaphyseal
One side more involved than other
Classification
Monostotic or polyostotic
McCune-Albright (Albright’s) syndrome is:
Polyostotic fibrous dysplasia
Café-au-lait spots (“coast of Maine” irregular border)
Precocious puberty or other endocrine abnormalities (Box 7.3-1)
Mazabraud’s syndrome
Fibrous dysplasia
Soft tissue myxomas
Box 7.3-1 Conditions Associated with Mccune-Albright Syndrome
Sexual precocity
Pituitary adenoma
Hyperthyroidism
Gastrointestinal polyps
Thymus hyperplasia
Splenic hyperplasia
Pancreatic islet cell hyperplasia
Hepatobiliary disease
Cardiac disease
Failure to thrive
Metabolic acidosis
Abnormalities in serum electrolytes, glucose, or insulin levels
Hyperphosphaturic hypophosphatemia
Osteosarcoma
Developmental delay
Microcephaly
Sudden or premature death
Diagnosis
Clinical Features
Polyostotic Fibrous Dysplasia
May be asymptomatic, although that is less common in polyostotic form
Pain
Limp (can be from pain, leg-length discrepancy, or Trendelenburg gait from “shepherd’s crook” deformity of proximal femur)
Swelling (if bone in subcutaneous location)
Angular deformity
Leg-length discrepancy
50% with craniofacial manifestations
McCune-Albright Syndrome (features in addition to the above)
Cutaneous macular pigmentation similar to café-au-lait spots of neurofibromatosis
Irregular “coast of Maine” border (unlike smooth “coast of California” border seen in neurofibromatosis) (Fig. 7.3-1)
Tend to cluster centrally, especially on the back
Most frequent extraskeletal manifestation (approximately one third)
Unusual in monostotic fibrous dysplasia
Precocious puberty or endocrinopathy
Precocious puberty more common (20%)
Females > males
Female presentation: vaginal bleeding, premature development of sexual organs, premature secondary sex characteristics
Male presentation: enlarged genitals, premature secondary sex characteristics
Endocrinopathy may include hyperparathyroidism, hyperthyroidism, Cushing syndrome, acromegaly, diabetes, rickets, osteomalacia, hyperprolactinemia.
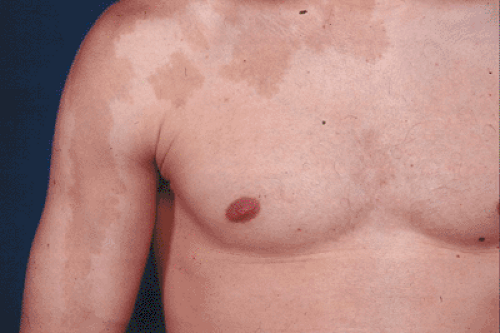 Figure 7.3-1 Irregular “coast of Maine” border in the pigmented skin lesion associated with polyostotic fibrous dysplasia. This differs from the smooth “coast of California” border seen typically in type I peripheral neurofibromatosis and in Jaffe-Campanacci syndrome (multiple nonossifying fibromas and café-au-lait spots). |
Mazabraud’s Syndrome
Myxomas with fibrous dysplasia bone lesions
Usually polyostotic (86%) over solitary fibrous dysplasia, rarely with McCune-Albright’s
Radiologic Features
Diaphyseal or metaphyseal
Epiphyseal involvement rare
Involvement of flat bones (skull, jaw, ribs) common
Spinal involvement uncommon
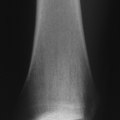
Lucent or “ground glass” appearance (Fig. 7.3-2)
May have calcifications
Sclerotic rim typical
May expand cortex (usually does not break cortex)
May have angular deformities (e.g., “shepherd’s crook” deformity) (Fig. 7.3-3)
Increased uptake on bone scan
Histologic Features
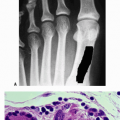
Fibrous stroma with spindle cells
Spicules of osteoid or woven bone that have the appearance of “alphabet soup” (often described as resembling the letters C, O, J, and Y) or “Chinese characters” (Fig. 7.3-4)
Few osteoblasts (lacking osteoblastic rimming)
Lack of osteoblastic rimming distinguishes fibrous dysplasia from osteofibrous dysplasia, more common in the tibia, which is characterized by osteoblastic rimming.
Cartilaginous foci may be present.
Treatment
Observe if asymptomatic. Endocrinologist should manage precocious puberty or other endocrinopathies.
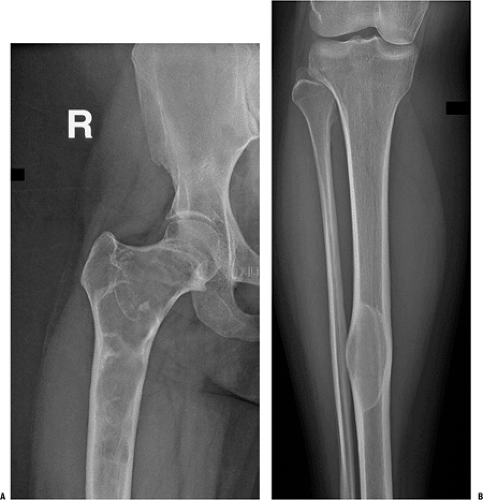 Figure 7.3-2 Radiographs from a patient with polyostotic fibrous dysplasia show extensive involvement of the proximal femur (A) and a single lesion in the ipsilateral tibia (B). Note the characteristic loss of normal trabeculation within the lesion in the tibia, which has been referred to as a “ground glass” mineralization pattern. This less organized appearance reflects the histology, which shows a more random “Chinese character” pattern of mineralization. |
Indications for Surgery
Biopsy indicated if diagnosis in question
Fracture through lesion
If alignment unacceptable or high-risk location (e.g., proximal femur)
In children may be able to treat pathologic fractures with casting
Progressive deformity
If causing functional disability, unacceptable disfigurement, significantly increased risk of pathologic fracture
Pain
Surgical Technique
In children, simple curettage or curettage and bone grafting typically are followed by recurrence of the lesion.
Prophylactic intramedullary nails may be useful in the setting of progressive deformity (see Fig. 7.3-3).Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree