Secondary Forms of Diabetes
Om P. Ganda
The term secondary diabetes generally refers to diabetes or glucose intolerance that develops in association with disorders (or factors) other than those currently defined as type 1 or type 2 diabetes mellitus or gestational diabetes mellitus. According to the classification system developed by the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus (1), the subclass “secondary diabetes” contains a variety of types of diabetes, in some of which the etiologic relationship is known (e.g., diabetes secondary to pancreatic disease, endocrine disease, or administration of certain drugs). In others, an etiologic relationship is suspected because of a higher frequency of association of diabetes with a syndrome or condition (e.g., a number of genetic syndromes). The extent of glucose intolerance in patients with secondary forms of diabetes varies widely, presenting as insulin-requiring or non-insulin-requiring overt diabetes, simulating type 1 (insulin-dependent diabetes mellitus) or type 2 diabetes (non-insulin-dependent diabetes mellitus) or as milder forms such as impaired fasting glucose (IFG), impaired glucose tolerance (IGT), or minimally abnormal glucose tolerance, considered nondiagnostic. Another complexity in the evolution of secondary diabetes is that an underlying coexisting predisposition to primary diabetes might be unmasked, a not uncommon occurrence, considering the prevalence of diabetes and the presumed genetic underpinnings of diabetes in the population.
Table 27.1 presents a classification of various forms of secondary diabetes. When diabetes is secondary to pancreatic disorders, particularly when β-cell mass is greatly reduced, e.g., by malignancy or pancreatectomy, or when diabetes is due to chemical agents toxic to the β-cell, e.g., pentamidine or pyriminil (Vacor), overt diabetes with or without ketoacidosis often will result, depending on the extent of β-cell loss. On the other hand, when diabetes is secondary to endocrinopathies leading to counterregulatory hormone production (e.g., acromegaly, Cushing syndrome, hyperthyroidism), overt diabetes, or ketoacidosis are unusual, thanks to the capacity of the β-cell reserve (2,3). Thus, the net metabolic outcome in patients with secondary diabetes depends on the direct or indirect impact of the underlying disorders on (a) insulin secretion, i.e., inhibition or compensatory hyperinsulinemia; (b) insulin sensitivity, i.e., glucose utilization; and (c) hepatic glucose output.
TABLE 27.1. A Classification of Secondary Forms of Diabetes or Impaired Glucose Tolerance | ||||
|---|---|---|---|---|
|
PANCREATIC DIABETES
Pancreatectomy
Diabetes that results from pancreatectomy is, in a mechanistic sense, the prototype of insulin-deficient secondary diabetes. The amount of human pancreas that must be surgically removed or pathologically destroyed before the development of fasting hyperglycemia is a matter of some debate. Of interest, elegant studies in baboons revealed that after induction of diabetes with streptozotocin, the animals developed fasting hyperglycemia and a reduction in β-cell function in vivo when 40% to 50% of the β-cell mass was still detectable by islet morphometric assessment (4). Furthermore, of 28 healthy human donors who underwent 50% pancreatectomy, 7 (25%) developed glucose intolerance and a deterioration in insulin secretion after 8 to 15 months (5) (Fig. 27.1). However, none of these individuals developed overt diabetes during this time. It has been suggested that development of diabetes in partially pancreatectomized humans depends on several additional factors, such as rate of regeneration of β-cells, changes in nutritional status caused by weight loss and concomitant exocrine insufficiency, and the glucagon deficiency resulting from loss of α-cells (6).
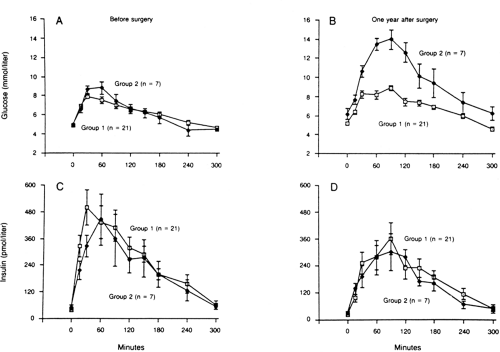 Figure 27.1. Mean (± SEM) serum glucose levels (A, B) and serum insulin levels (C, D), measured before and 1 year after hemipancreatectomy during 5-hour oral glucose tolerance tests in 21 transplant donors with normal glucose tolerance at 1 year (group 1; open squares) and 7 donors with abnormal glucose tolerance at 1 year (group 2; closed circles). (From Kendall DM, Sutherland DER, Najarian JS, et al. Effects of hemipancreatectomy on insulin secretion and glucose tolerance in healthy humans. N Engl J Med 1990;322:898–903, with permission. Copyright © 1990 Massachusetts Medical Society. All rights reserved.) |
Subtotal or total pancreatectomy theoretically provides a model for diabetes with “pure” insulin deficiency, because glucagon also should be absent (7). However, there are many reports of normal or elevated levels of immunoreactive glucagon (IRG) originating from extrapancreatic sources (gastrointestinal) in such patients (8,9,10). In most studies, the majority of this IRG comprised larger-molecular-weight material, which does not have biologic activity, but small quantities of 3500-kDa pancreatic glucagon may also be detectable (8). The biologic significance of extrapancreatic glucagon in these patients is not known, and their levels of IRG do not respond to arginine administration, although paradoxical stimulation in response to oral glucose or mixed meals has been reported (8,10).
The clinical presentation of diabetes in pancreatectomized humans differs from that of type 1 diabetes in several respects:
Because of coexisting exocrine deficiency, pancreatectomized individuals present a nutritional challenge. They tend to be leaner than patients with type 1 diabetes and present with various degrees of malabsorption despite treatment with pancreatic enzyme supplements. Consequently, their insulin requirements are lower than those of weight-matched patients with type 1 diabetes (11).
Pancreatectomized patients are more prone to hypoglycemia and may have a more sluggish and delayed recovery from hypoglycemia. The reasons for these differences are multiple. Glucose turnover and gluconeogenesis are considerably diminished, and levels of gluconeogenic precursors (alanine, lactate, pyruvate) are markedly increased owing to lack of glucagon (12,13); these changes are reversible by physiologic replacement of glucagon. Furthermore, epinephrine release in response to hypoglycemia may be markedly blunted (14), possibly due to recurrent episodes
of hypoglycemia. Thus, combined defects in glucagon and epinephrine may underlie prolonged and delayed recovery from hypoglycemia in such patients.
Ketonemia and ketoacidosis are less severe in pancreatectomized patients despite comparable stimulation of lipolysis. As shown in Figure 27.2, withdrawal of insulin for 12 hours from totally pancreatectomized subjects (n = 4) and age- and weight-matched patients with type 1 diabetes (n = 6) resulted in about a 50% reduction in the magnitude of hyperglycemia and hyperketonemia in the former (15). There were no significant differences in levels of free fatty acids and glycerol. Glucagon was undetectable before and after insulin withdrawal in pancreatectomized subjects (7,15). These studies strongly support the important role of glucagon in the promotion of ketogenesis but also argue against a primary role of glucagon in the induction of ketoacidosis.
The issue of insulin sensitivity and peripheral glucose utilization following pancreatectomy is controversial. Although some investigators reported increased cellular insulin binding and enhanced hepatic and extrahepatic insulin sensitivity (16), others observed a state of peripheral insulin resistance (17) that was further impaired by glucagon replacement (18). Some of these discrepancies may be explained by differences in patient characteristics and alcohol intake and by the lack of consistent control for the induction of insulin resistance by antecedent hyperglycemia per se.
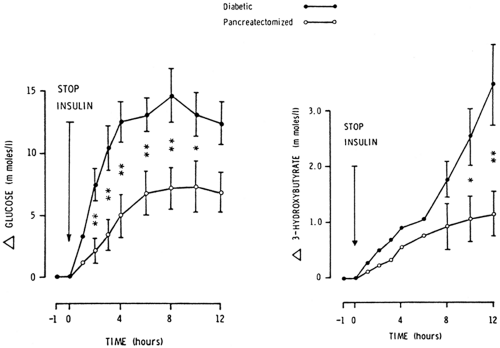 Figure 27.2. Changes in blood concentrations (mean ± SEM) of glucose and 3-hydroxybutyrate in patients with type 1 diabetes (n = 6) and pancreatectomized subjects (n = 4) after withdrawal of insulin. * P > 0.05; ** P > 0.02. (From Barnes AJ, Bloom SR, Alberti KGMM, et al. Ketoacidosis in pancreatectomized man. N Engl J Med 1977;296:1250–1253, with permission. Copyright © 1977 Massachusetts Medical Society. All rights reserved.) |
Pancreatitis
Diabetes secondary to pancreatitis accounts for fewer than 1% of cases of diabetes in the United States and other Western countries. However, in many parts of the world (especially tropical countries), the proportion is much higher.
Transient hyperglycemia may be seen in up to 50% of patients with acute pancreatitis in the absence of further attacks, but persistent diabetes develops—presumably as a result of ongoing chronic, painless pancreatitis—in fewer than 5% of such individuals during long-term follow-up in the absence of further attacks (19). On the other hand, in patients with chronic, relapsing pancreatitis, the incidence of diabetes increases over the years, being present in about 40% to 50% of patients after 20 years, with an additional 25% to 30% having IGT (19,20). Among patients with fibrocalcific pancreatitis, up to 80% to 90% have overt diabetes or IGT (19,20). In Western societies, the etiology of chronic pancreatitis is largely alcohol related in both patients with and without biliary disease. The precise mechanisms by which chronic pancreatic inflammation leads to glucose intolerance are not established, but compromised blood flow to islets from fibrotic scarring of exocrine pancreas may play a role. Insulin and C-peptide secretory responses to various secretagogues, including orally and intravenously administered glucose, sulfonylureas, glucagon, and amino acids, are impaired, and these abnormalities are correlated with the magnitude of exocrine pancreatic dysfunction (19,21,22). Glucagon levels are markedly increased in acute pancreatitis, both in the basal state and following stimulation with alanine. This increase may contribute to the transient hyperglycemia frequently seen in this situation (23). In chronic pancreatitis, the basal glucagon levels are normal or elevated, but the responses to amino acids (9,23) or to insulin-induced hypoglycemia (24,25) usually are found to be blunted. In some studies, however, levels of glucagon-like immunoreactivity were increased after stimulation (20,26), perhaps due to glucagon derived from extrapancreatic sources, but the significance of this observation is uncertain. Finally, in a small series of patients with chronic pancreatitis, a partial hepatic insulin resistance was shown in the presence of pancreatic polypeptide (PP) deficiency, which was reversible by PP infusion (27).
Patients with diabetes secondary to chronic pancreatitis may show a delayed recovery from hypoglycemia, a situation similar to that seen in pancreatectomized subjects. However, the incidence and severity of hypoglycemia in these patients are influenced by a number of other factors, e.g., alcohol intake, nutritional status, and state of malabsorption.
Subtle abnormalities of pancreatic exocrine function may sometimes be seen in patients with type 1 or type 2 diabetes (28,29), a finding that suggests the loss of a trophic effect of islet cell secretion on acinar cell function. These abnormalities, usually asymptomatic, may occasionally be difficult to differentiate from chronic, painless pancreatitis and may make additional diagnostic workup necessary.
Cystic Fibrosis
Cystic fibrosis (CF), one of the most common autosomal recessive disorders in the white population, leads to chronic progressive pulmonary disease and pancreatic insufficiency. With the increasing life expectancy of patients with CF, the incidence of diabetes due to CF has been increasing as a comorbidity. IGT or diabetes has been reported in up to 50% of adult patients with CF (30,31,32). Progressive insulin deficiency develops because of fibrotic destruction of islets and a marked decrease in β-cell mass (31). In addition, islet amyloid deposition was detected in 69% of 41 diabetic cases in one series (33). The extent of glucose intolerance depends also on additional factors, e.g., malnutrition, infections, and liver dysfunction. Recently, an increased prevalence of mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene was reported in patients with “idiopathic” chronic pancreatitis as well as “alcoholic” chronic pancreatitis in the absence of any diagnostic features of CF (34,35). In most of these patients, one abnormal allele was detected. Thus, heterozygosity for CFTR mutation might be an underlying defect in many adult patients presenting with chronic pancreatitis.
Pancreatic Cancer
Pancreatic malignancy, leading to loss of β-cell mass, is a well-known cause of diabetes. However, whether diabetes is a risk factor for pancreatic malignancy remains controversial. In some case-control studies, diabetes could not be shown to be a risk factor, after excluding patients with recent onset (>3 years) of hyperglycemia (36). However, in a large meta-analysis of 20 case-control or cohort studies, a twofold relative risk of pancreatic cancer was found, even after excluding those with diabetes of up to 5 years of duration (37). Furthermore, in a large, prospective cohort study, with a 25-year duration of follow-up, a progressive increase in the risk of pancreatic cancer was seen with increasing glucose levels following a glucose load at baseline (38). Whether hyperglycemia itself or the associated metabolic abnormalities, e.g., hyperinsulinemia or abnormalities in the insulin-like growth factor (IGF) axis, are involved in pancreatic carcinogenesis is unclear. In a series of patients with pancreatic cancer, with or without diabetes, levels of islet amyloid polypeptide (IAPP) were more than twice as high in those with diabetes compared with those with normal glucose tolerance (39).
HEMOCHROMATOSIS
Prevalence and Clinical Features
Hemochromatosis is the most common autosomal recessive disorder; the gene frequency is 7% to 10% among white populations, and the disease prevalence is 2 to 4 per 1,000 population (40,41,42,43,44). The disease is expressed three to five times more frequently in men than in women, since about 60% to 80% of homozygous women do not accumulate iron significantly because of menstrual blood loss and pregnancies.
The hemochromatosis gene (HFE) has been localized to chromosome 6, and two missense point mutations, C282Y and H63D, account for 65% to 100% of all patients of European descent (41,44). H63D is not deleterious by itself but probably acts synergistically in persons who are also heterozygotes for C282Y. In large series of participants, gene frequencies for C282Y and H63D mutations were 5% to 6% and 13% to 15%, respectively, whereas about 2% were compound heterozygotes. However, only about 50% of homozygotes for C282Y express clinical features of hemochromatosis, more commonly men than women (45,46).
Iron deposition occurs primarily in parenchymal cells of liver, pancreas, adrenals, anterior pituitary, myocardium, and skeletal muscle. The classic triad of hepatomegaly, diabetes, and skin pigmentation (“bronze diabetes”), once considered common, is in fact not a frequent association, considering the changing clinical presentation associated with early diagnosis and earlier treatment (41,45). Some of the common presenting symptoms are hepatomegaly with or without abdominal pain, arthralgias, fatigue, and impotence. Presence of symptoms usually correlates with the severity of iron accumulation as documented by liver biopsy and with the presence of cirrhosis. Cirrhosis was present at diagnosis in about 70% of patients prior to the availability of genetic tests (47). Hepatocellular carcinoma develops in about 15% to 30% of patients, depending on patients’ longevity, despite the successful removal of iron.
Hemochromatosis may have a number of secondary causes, including sideroblastic anemias (chiefly thalassemia major), chronic hemolytic anemias, multiple blood transfusions, porphyria cutanea tarda, and dietary or medical iron overload, e.g., consumption of iron-rich beer among the Bantus (40,41). None of these conditions is linked with HFE gene mutations (41). The severity of total-body iron load produced by these states is variable but is usually less than the loads of 30 g or more seen in primary (idiopathic) hemochromatosis. Alcohol promotes iron absorption but does not, by itself, result in hemochromatosis.
Diabetes in Hemochromatosis
Abnormal glucose tolerance occurs in up to 75% to 80% of patients with hemochromatosis; of these, 50% to 60% have overt diabetes (45,47,48). Similarly, glucose intolerance was present in about 50% of patients with thalassemia major following chronic transfusion therapy (49,50). In a large series of patients with hemochromatosis, 25% had first-degree relatives with diabetes (48).
Conversely, hemochromatosis was diagnosed in 1.3% of 894 patients with diabetes by the old criteria in Italy, where the gene frequency is lower (51).The prevalence in age- and sex-matched controls was 0.2%. The pathogenesis of glucose intolerance in iron-overload states remains controversial, because multiple factors, including cirrhosis, pancreatic iron deposition, and underlying primary diabetes, can be involved. However, the severity of cirrhosis and iron load is correlated with the degree of glucose intolerance, and the control of diabetes improves in 35% to 45% of all patients following iron depletion (40,47).
Studies of β-cell and α-cell function in the presence of iron overload have revealed several interesting features. Patients who develop overt diabetes usually have impaired β-cell function (48,49), and about 40% to 50% of them require insulin therapy. However, in patients studied in a precirrhotic stage
and in those who have not developed overt diabetes, significant hyperinsulinemia was observed in response to oral glucose (52,53) and during a hyperglycemic clamp (53). Furthermore, rates of insulin-mediated glucose disposal were impaired in patients with transfusion-induced iron overload in the latter study (53). These observations suggest that insulin resistance secondary to hepatic or extrahepatic (muscle?) iron deposition precedes the β-cell dysfunction and overt diabetes that eventually develop. However, the mechanism of insulin resistance produced by iron overload remains unknown. In histopathologic studies, an increase in β-cell mass was demonstrated in a small number of nondiabetic or mildly diabetic patients with iron overload (54). However, whether iron infiltration of pancreatic islets explains the later β-cell dysfunction remains controversial. In patients with hemochromatosis, glucagon secretion was augmented by arginine and was not suppressed by oral glucose (55,56), responses similar to those seen in primary diabetes. In this respect, the α-cell responses seem to differ from those seen in patients with chronic pancreatitis (21,22) and are in keeping with the observation that iron deposition in islets, albeit variable, is restricted to β-cells (54).
and in those who have not developed overt diabetes, significant hyperinsulinemia was observed in response to oral glucose (52,53) and during a hyperglycemic clamp (53). Furthermore, rates of insulin-mediated glucose disposal were impaired in patients with transfusion-induced iron overload in the latter study (53). These observations suggest that insulin resistance secondary to hepatic or extrahepatic (muscle?) iron deposition precedes the β-cell dysfunction and overt diabetes that eventually develop. However, the mechanism of insulin resistance produced by iron overload remains unknown. In histopathologic studies, an increase in β-cell mass was demonstrated in a small number of nondiabetic or mildly diabetic patients with iron overload (54). However, whether iron infiltration of pancreatic islets explains the later β-cell dysfunction remains controversial. In patients with hemochromatosis, glucagon secretion was augmented by arginine and was not suppressed by oral glucose (55,56), responses similar to those seen in primary diabetes. In this respect, the α-cell responses seem to differ from those seen in patients with chronic pancreatitis (21,22) and are in keeping with the observation that iron deposition in islets, albeit variable, is restricted to β-cells (54).
In making the diagnosis of hemochromatosis, it should be noted that biochemical tests of iron metabolism, i.e., determinations of serum iron, transferrin saturation, and ferritin levels, are helpful but are not as specific for the diagnosis as is the hepatic histology (41).The availability of genetic tests helps diagnosis in relatives at early stages (45).Recently, attention was drawn to the presence of hyperferritinemia in 10% to 30% of unselected patients with type 2 diabetes (57). In most cases, the hyperferritinemia in such patients resolved spontaneously or proved to be due to other causes when follow-up studies were done (58).An underlying chronic inflammatory state also may result in a nonspecific increase in iron stores in patients with insulin resistance; the relevance of this to development of diabetes and its complications needs further study (59). Such studies underscore the need for confirmation of results, particularly when patients with diabetes are being screened for hemochromatosis.
Management and Prognosis
Regular phlebotomy treatment, often lifelong, remains the treatment of choice for hemochromatosis. Successful iron depletion, initiated early in the course of disease, clearly reduces the incidence and progression of cirrhosis, improves diabetes control, and frequently reduces damage to other target organs and overall morbidity and mortality (41,47). In a large prospective study of 163 patients, with a mean follow-up period of 10.5 ± 5.6 years, the major determinants of reduced survival included the presence of cirrhosis, diabetes, and failure of iron depletion (47) (Fig. 27.3). Mortality ratios (observed/expected) for liver cancer, cardiomyopathy, cirrhosis, and diabetes were 219, 306, 13, and 7, respectively, for patients with hematochromatosis as compared with those for the general population in this study. Successful iron depletion, however, did not protect from the development of liver cancer.
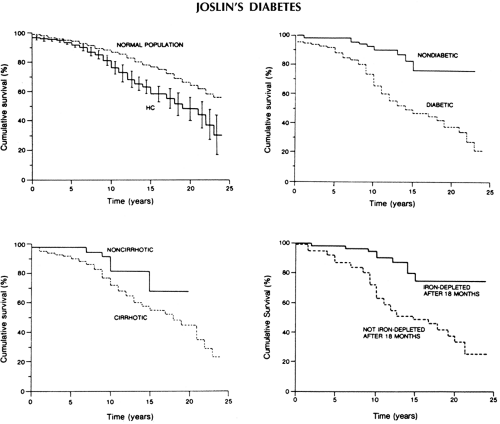 Figure 27.3. Cumulative survival in 163 patients with hemochromatosis compared with normal population and in the same patients with (n = 112) or without (n = 51) cirrhosis; with (n = 89) or without (n = 74) diabetes; and depleted (n = 77) or not depleted (n = 75) of iron during the first 18 months of venesection (see text for details). All differences were statistically significant (P > 0.05 to > 0.002 by log-rank test). (From Niederau C, Fischer R, Sonnenberg A, et al. Survival and causes of death in cirrhotic and noncirrhotic patients with primary hemochromatosis. N Engl J Med 1985;313:1256–262, with permission. Copyright © 1985 Massachusetts Medical Society. All rights reserved.) |
ENDOCRINOPATHIES
The major sites of action of various counterregulatory hormones on target organs and the principal mechanisms of their diabetogenic effects are listed in Table 27.2.
TABLE 27.2. Sites of Actions of Major Diabetogenic Hormones in Humans | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Disorders of Growth Hormone Secretion
Classic experiments, using crude pituitary extracts, performed more than 60 years ago demonstrated a relationship between the anterior pituitary and diabetes (62). In rat pancreatic monolayer cultures derived from an islet tumor cell line (63) or neonatal pancreas (64), growth hormone (GH) stimulated β-cell replication, an effect that was claimed to be independent of GH-induced insulin-like growth factors (64). However, chronic administration of GH in experimental animals over prolonged periods has been shown to result in hypersecretion of insulin followed by eventual loss of β-cells and permanent diabetes (65).
SECRETION AND METABOLIC EFFECTS OF GROWTH HORMONE
The physiology of GH secretion and its diverse metabolic effects on carbohydrate, protein, and lipid metabolism in humans have been studied extensively (66,67,68). In healthy subjects, GH release is stimulated by hypoglycemia, sleep, exercise, stress, and amino acids and is inhibited by hyperglycemia. Recently, a 28-amino-acid gastric hormone, ghrelin, has been identified as a potent ligand for GH secretagogue receptor (GHS-R), probably mediating GH secretion in response to various stimuli via neurons in the hypothalamus secreting growth hormone-releasing hormone (GHRH) (69). Infusions of GH in healthy subjects that produce increments in plasma GH levels within the supraphysiologic range (up to 30 to 50 ng/mL) initially result in acute but transient (2 to 4 hours) insulin-like effects, i.e., suppression of hepatic glucose production and enhancement of peripheral glucose clearance (69,70,71,72). In fact, insulin levels increase over this period, offering an explanation for some portion of these effects. Subsequently, over a 2- to 12-hour period, similar increments or increments closer to the physiologic range (73) are associated with the “delayed” insulin-antagonistic effects. The primary site of GH-induced insulin resistance resides at the level of peripheral tissues (72,73), as first suggested by the elegant studies of forearm perfusion by Zierler and Rabinowitz (74). Although an effect of GH on inhibition of peripheral glucose uptake is well established, the lipolytic effect has not always been confirmed (67,75).
The understanding of the cellular site of insulin resistance induced by GH is complicated; multiple coexisting factors are involved, including GH concentrations, GH-binding proteins, presence or absence of hyperinsulinemia, and glucose intolerance. In most studies of healthy subjects given GH infusions or of patients with acromegaly, binding of insulin to receptors on circulating monocytes or hepatocytes was found to be little affected as a result of reciprocal effects on receptor number and affinity (72,73,66,76). However, in acromegalic patients with fasting hyperglycemia, the compensatory increase in receptor affinity that occurs in such patients with normoglycemia may fail to occur, a factor contributing to glucose intolerance (77). In cultured adipocytes, GH was shown to inhibit the expression of the gene for the glucose transporter GLUT1 but not that for the transporter GLUT4 (78). Thus, probably both postreceptor and receptor defects in insulin action underlie the diabetogenic effects of GH excess, although the precise nature of these defects remains unknown.
DIABETES IN ACROMEGALY
Anterior pituitary adenomas that hypersecrete GH account for more than 90% of cases of acromegaly (79). Other causes include ectopic (nonpituitary) sources of GH or GHRH, e.g., pancreatic islet cell tumors, carcinoid tumors, and hypothalamic hamartomas.
Glucose intolerance is prevalent in patients with acromegaly, affecting about 60% to 70%. However, overt diabetes requiring treatment occurs in only 10% to 30% of patients (80,81,82,83). Even more frequent than glucose intolerance is evidence for insulin resistance, which is manifested by a striking hyperinsulinemia in response to orally or intravenously administered glucose and other secretagogues, as well as markedly attenuated responses to exogenous insulin (67,68,84). Defects in both hepatic action and peripheral glucose disposal have been described in studies using insulin clamp techniques (70,73).Overall, GH levels in individual patients correlate poorly with both hyperinsulinemia and the severity of glucose intolerance. A better correlation of disease activity and glucose intolerance is found with serum levels of insulin-like growth factor 1 (IGF-1) (67,68) or
IGF-binding protein 3 (IGFBP-3) (85). In contrast to patients with acromegaly with normal or impaired glucose tolerance, insulin secretion is quite blunted by the time overt diabetes develops, with or without ketosis (68,84), a finding reminiscent of that in animal models of metasomatotrophic diabetes (65). However, coexistent primary diabetes (type 1 or type 2) may also be present, particularly if diabetes persists after successful treatment of acromegaly in such patients. Considerable evidence exists from studies of somatostatin infusion that lipolytic and ketogenic effects of GH supervene only after significant insulinopenia (71,86). Successful treatment of acromegaly, with normalization of GH and IGF-1, is usually accompanied by striking improvements in glucose tolerance and a reversal of hyperinsulinemia, as well as by normalization of insulin sensitivity (68,82,83,84). However, the results are unpredictable in those with overt, symptomatic diabetes (84,87). Lack of a complete remission in many patients may be due to incomplete cure of acromegaly, defined as suppression of GH to less than 2.5 ng/mL or to less than 1 ng/mL after oral glucose, using newer, ultrasensitive assays (88). Some have argued that this cut-off should be even lower, i.e., less than 0.5 ng/mL (89). The increase in mortality rate in acromegaly may persist unless GH levels are suppressed to the near-normal range (90,91).Moreover, glucose intolerance and hypertension are independent determinants of acromegalic cardiomyopathy (92).
IGF-binding protein 3 (IGFBP-3) (85). In contrast to patients with acromegaly with normal or impaired glucose tolerance, insulin secretion is quite blunted by the time overt diabetes develops, with or without ketosis (68,84), a finding reminiscent of that in animal models of metasomatotrophic diabetes (65). However, coexistent primary diabetes (type 1 or type 2) may also be present, particularly if diabetes persists after successful treatment of acromegaly in such patients. Considerable evidence exists from studies of somatostatin infusion that lipolytic and ketogenic effects of GH supervene only after significant insulinopenia (71,86). Successful treatment of acromegaly, with normalization of GH and IGF-1, is usually accompanied by striking improvements in glucose tolerance and a reversal of hyperinsulinemia, as well as by normalization of insulin sensitivity (68,82,83,84). However, the results are unpredictable in those with overt, symptomatic diabetes (84,87). Lack of a complete remission in many patients may be due to incomplete cure of acromegaly, defined as suppression of GH to less than 2.5 ng/mL or to less than 1 ng/mL after oral glucose, using newer, ultrasensitive assays (88). Some have argued that this cut-off should be even lower, i.e., less than 0.5 ng/mL (89). The increase in mortality rate in acromegaly may persist unless GH levels are suppressed to the near-normal range (90,91).Moreover, glucose intolerance and hypertension are independent determinants of acromegalic cardiomyopathy (92).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


