SARS-CoV-2 and Coronaviruses
Evolutionary Origins
In the last two decades, three coronaviruses have caused outbreaks of varying scales, with the pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) representing the most recent threat to human health at a global level. Aside from severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), which were responsible for the first two outbreaks of the 21st century, only four other coronaviruses that cause relatively mild disease in humans have been discovered: HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1. , The SARS, MERS, and COVID-19 outbreaks have demonstrated that zoonotic coronaviruses can successfully cross species barriers to infect humans and cause a high level of pathogenicity and mortality.
SARS-CoV-2 belongs to the order Nidovirales, family Coronaviridae, subfamily Coronavirinae, and genus Betacoronavirus. Within the genus Betacoronavirus, four distinct lineages are assigned: HCoV-OC43 and HCoV-HKU1 belong to lineage A, SARS-CoV and SARS-CoV-2 belong to lineage B, and MERS-CoV belongs to lineage C. SARS-CoV-2 is further classified under the subgenus Sarbecovirus. , SARS-CoV and MERS-CoV both originated in bats, palm civets acted as the intermediate host for SARS-CoV, and camels served as the intermediate host for MERS-CoV. The genome of SARS-CoV-2 shares 80% sequence identity with SARS-CoV and presents a high degree of sequence identity to the genomes of bat coronaviruses RaTG13 and RmYN02. , The high level of similarity to bat-derived coronaviruses suggests that SARS-CoV-2 must have also originated in bats. , Although sarbecoviruses are known to undergo frequent recombination, assessment of the SARS-CoV-2 genome revealed no evidence to suggest that it originated from a recent recombination event. Early studies characterizing SARS-CoV-2 reported that it uses the same human receptor as SARS-CoV, angiotensin-converting enzyme-2 (ACE2), to enter and infect host cells. In contrast, MERS-CoV uses a receptor called dipeptidyl peptidase 4 (DPP4). Interestingly, SARS-CoV-2 possesses a polybasic cleavage site insertion (PRRA sequence) in the spike protein at the junction of the S1 and S2 subunits, which resembles a sequence that is present in MERS-CoV but absent in SARS-CoV and RaTG13. This sequence was identified as a putative furin cleavage site that may be acted upon by the proprotein convertase furin during viral egress.
The discovery of a coronavirus similar to SARS-CoV-2, pangolin CoV, in Malayan pangolins showing clinical signs of infection drove the suspicion that pangolins may serve as the intermediate host for SARS-CoV-2. The receptor-binding domain (RBD) in the spike protein of pangolin-CoV was almost identical to that of SARS-CoV-2, but pangolin-CoV lacked the furin cleavage site found in SARS-CoV-2. Moreover, many bat-derived coronaviruses, such as RmYN02, have been reported to contain a similar insertion in spike protein at the S1/S2 junction, suggesting that SARS-CoV-2 likely originated from multiple recombination events that occurred within viruses inhabiting bats and other species. With the accumulating evidence, it appears unlikely that pangolins acted as intermediate hosts facilitating SARS-CoV-2 spillover to humans. , Although the involvement of an intermediate host cannot be ruled out, it has been suggested that SARS-CoV-2 may have spilled over directly from bats to humans without requiring an intermediate host. The origin and cross-species transmission of SARS-CoV-2 is still under investigation and remains a subject of debate.
Structure, Genome, and Proteome
The positive-sense single-stranded RNA genome of SARS-CoV-2 extends to 29,891 nucleotides and encodes 9860 amino acids. In addition to the flanking 5′ and 3′ untranslated regions (UTRs), the SARS-CoV-2 genome includes coding regions for the structural proteins spike glycoprotein (S), envelope protein (E), membrane glycoprotein (M), and nucleocapsid protein (N), and several open reading frames (ORF1ab, ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9b, and ORF10) that code for accessory structural and nonstructural proteins ( Fig. 2.1 ).
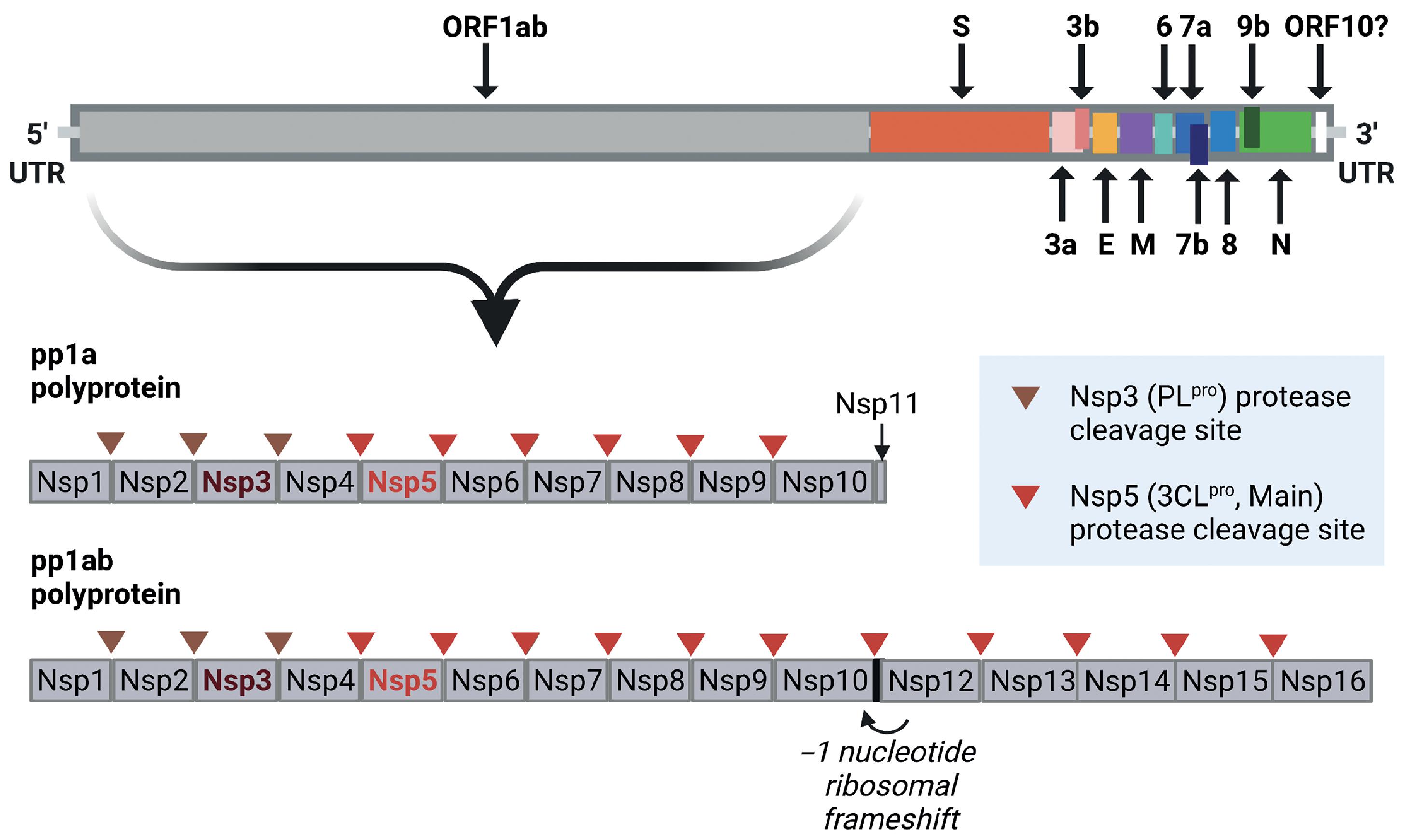
The first electron micrographs of SARS-CoV-2 revealed virus particles described as spherical with some pleomorphism and distinctive spikes that created the appearance of a solar corona. Coronavirus particles consist of an outer lipid bilayer envelope inserted with the spike, membrane, and envelope structural proteins. The inner core contains a helical nucleocapsid structure that is formed by the association of nucleocapsid phosphoproteins with the viral genomic RNA ( Fig. 2.2 ).
Structural Proteins
Spike Protein
The spike glycoprotein (S) is a structural protein that is critical for binding to the ACE2 receptor on host cells and facilitating cell entry. The S protein is embedded into the outer lipid bilayer envelope in a uniform distribution and extends out from the virion surface, producing the defining “corona”-like appearance. The SARS-CoV-2 S protein is a densely glycosylated homotrimeric class I fusion protein that is divided into two subunits, S1 and S2, which are separated by a multibasic furin protease cleavage site. The S1 subunit forms a globular head that binds to the host cell receptor, and the S2 subunit facilitates viral membrane fusion with the host cell membrane. The S1 subunit is composed of an amino- or N-terminal domain (NTD) and a carboxy- or C-terminal domain (CTD). The S1 subunit CTD functions as the RBD that binds to the human ACE2 (hACE2) receptor. ,
The S protein exists in a metastable prefusion conformation. On binding to the host cell receptor, it undergoes a significant structural transformation to enable fusion of the viral membrane with the host cell membrane. The furin cleavage site (RRXR motif) at the S1/S2 junction is proteolytically cleaved, leading to the separation of S1 from S2; however, the two subunits remain noncovalently bound to each other. , After S1/S2 cleavage and engagement of the RBD with the host cell ACE2 receptor, another cleavage site (S2′) in the S2 subunit is exposed, and this site also must be cleaved by proteases to release the fusion peptide, which is essential for membrane fusion and viral infectivity. Receptor binding destabilizes the prefusion trimer, resulting in the shedding of the S1 subunit and transition of the S2 subunit to a stable postfusion conformation.
To engage the host cell receptor, the RBD in the S1 subunit undergoes hinge-like structural movements that either expose or hide the residues involved in receptor recognition. The accessible state is referred to as the “open” or “up” conformation, and the inaccessible state is referred to as the “closed” or “down” conformation ( Fig. 2.3 ). The RBD consists of a core and a receptor binding motif (RBM), with the latter directly mediating contacts with the ACE2 receptor. Various studies have reported either equivalent or higher binding affinity of the SARS-CoV-2 RBD for ACE2, compared with its counterpart in SARS-CoV. , ,
Envelope Protein
The envelope glycoprotein (E) in SARS-CoV-2 is an integral membrane protein of only 75 amino acids in length. Envelope proteins are viroporins, which are generally described as virally encoded small proteins that can form pores in the membranes of host cell organelles and modulate ion channel activity, among other functions. In lipid bilayers that mimic the endoplasmic reticulum–Golgi intermediate compartment (ERGIC) membrane, the SARS-CoV-2 E protein transmembrane domain forms a five-helix bundle that surrounds a narrow pore, a homopentameric cation channel. The elucidated N-terminal lumen/C-terminal cytoplasmic membrane topology of SARS-CoV-2 E is conducive to the conduction of Ca 2+ ions out of the ERGIC lumen, a role that may link the E protein to host inflammasome activation based on similar topology and involvement of the SARS-CoV E protein in this process. , Examination of E protein from other coronaviruses indicates that the protein is abundantly expressed in infected cells, but only a small proportion of the protein is incorporated into the virion envelope. The larger proportion is localized at sites of intracellular trafficking, such as the endoplasmic reticulum (ER), Golgi, and ERGIC membranes. At these sites, the E protein plays a role in virion assembly and budding.
Membrane Protein
The most abundant protein in coronaviruses, the Membrane protein (M), is a transmembrane glycoprotein that plays a role in delineation of the viral envelope shape and size. The M protein acts as a scaffold to regulate virion assembly by binding to other viral structural proteins at the site of budding and bringing them together to form the viral envelope. The M protein is functionally dimeric and may be able to associate with other M dimers to form a matrix-like layer. The M protein network possesses intrinsic membrane-bending properties, but its interactions with the S protein, E protein, and N protein–viral RNA complexes are important for effective membrane curvature and has an impact on virion size. , The M protein interacts with the spike protein to facilitate its retention in the ERGIC/Golgi complex. The M protein can adopt two conformations; the elongated conformer of M protein plays a role in spike incorporation into new virions. The structural properties of this conformer facilitate the formation of a rigid and convex viral envelope. The C-terminal domain of the M protein further interacts with and stabilizes the internal core and N protein–RNA complexes that make up the nucleocapsid, to promote completion of virion assembly. Expression of the M, S, and E proteins is minimally required for the production of SARS-CoV-2 noninfectious virus-like particles.
Nucleocapsid Protein
The nucleocapsid (N) protein is a multivalent RNA-binding protein, the primary role of which is to package the 30-kb-long genomic RNA compactly into viral ribonucleoprotein (vRNP) complexes that can be accommodated into the approximately 80-nm-diameter viral lumen. The N protein self-associates and naturally exists in a dimeric state, although it can also form oligomers, a property that is likely important for the formation of vRNPs. The protein consists of two globular domains, the NTD and the CTD, with the NTD containing RNA-binding sites that may interact with the viral genomic RNA packaging signal and the CTD forming a dimer with an RNA-binding groove to aid in vRNP assembly. The NTD and CTD are separated by a central, highly conserved, intrinsically disordered region containing a serine arginine (SR)–rich sequence with multiple phosphorylation sites that are targeted by cytoplasmic kinases to regulate the function of the N protein. The N protein also includes N-terminal and C-terminal intrinsically disordered regions; the latter and the CTD are both involved in M protein binding to anchor the vRNP complex to the inner surface of the viral envelope.
Structural analysis of SARS-CoV-2 viruses by cryoelectron tomography revealed that vRNPs associated with the viral envelope were stacked into cylindrical or helical filament-like assemblies. Efficient packing of the virus particles by N proteins could be accomplished through a “beads on a string” formation with viral RNA linking neighboring vRNPs. Another study determined that the vRNPs in SARS-CoV-2 existed in different arrangements based on the shape of the virions. In spherical virions, there was a higher incidence of membrane-proximal vRNPs packed internally against the envelope in a “hexon” formation. In ellipsoidal virions, membrane-free vRNPs were arranged like pyramids or in “tetrahedron” formations. However, in both arrangements, neighboring vRNPs were equally spaced apart, and in situ projection suggests that tetrahedrons might be able to assemble into hexons. Therefore the vRNP triangle presents a durable building block that can withstand environmental and mechanical stresses and allows for the adoption of various arrangements within the virion.
Accessory Proteins
ORF3a
SARS-CoV-2 ORF3a encodes a viroporin that shares 73% sequence identity with SARS-CoV ORF3a. ORF3a has been demonstrated to play a role in blocking autophagy and conferring viral escape from lysosomal destruction, a function reported to be unique to SARS-CoV-2 ORF3a and not observed in its SARS-CoV counterpart. , Additionally, ORF3a is involved in inflammasome activation, pyroptosis, and apoptosis induction. , ,
ORF3b
Because of the presence of premature stop codons, ORF3b is a short protein that is 22 amino acids in length, with no homology to its SARS-CoV counterpart, which is 154 amino acids long and known to function as an interferon (IFN) antagonist. As a result of the completely different sequences, SARS-CoV-2 ORF3b was originally predicted to lack any similarity in function to SARS-CoV ORF3b. However, this novel short protein has been demonstrated to be a potent IFN antagonist that can suppress type I IFN (IFN-I) induction even more effectively than SARS-CoV ORF3b. SARS-CoV-2–related viruses found in bats and pangolins encode a similar truncated ORF3b with IFN antagonist activity. The C-terminal region of SARS-CoV ORF3b contains a nuclear localization signal (NLS) that is lacking in SARS-CoV-2 ORF3b. Truncation of the C-terminus of SARS-CoV ORF3b enhances its IFN antagonist activity, suggesting that the NLS may impair its ability to block the nuclear translocation of IRF3, a transcription factor that induces IFN-β (IFNB1) expression.
ORF6
SARS-CoV-2 ORF6 shares only 66% sequence similarity with its counterpart in SARS-CoV, and this variation in sequence mainly occurs at the C-terminus. SARS-CoV ORF6 plays a key role in antagonizing IFN signaling, and its C-terminal tail is critical for this function. Despite the differences in sequence, SARS-CoV-2 ORF6 displays an equivalent ability to antagonize IFN signaling by blocking nuclear translocation of the transcription factors IRF3, STAT1, and STAT2. , By directly interacting with NUP98-RAE1 in the nuclear pore complex by its CTD, SARS-CoV-2 ORF6 has been reported to interfere with the docking of the karyopherin/importin complex. ORF6 also interacts with karyopherin α2 (KPNA2), presenting an alternative mechanism that could disrupt nuclear translocation of proteins involved in IFN signaling.
ORF7a
Similar to its SARS-CoV ortholog, SARS-CoV-2 ORF7a is a transmembrane protein of 121 amino acids, including an immunoglobulin-like (Ig-like) ectodomain and a hydrophobic transmembrane domain. The Ig-like domain is typically found in proteins that mediate cell adhesion or protein–protein binding. The Ig-like domain in SARS-CoV-2 ORF7a mediates interactions with CD14+ monocytes. Although SARS-CoV-2 ORF7a is structurally similar to SARS-CoV ORF7a, the latter does not interact efficiently with CD14+ monocytes. Additionally, ORF7a plays a role in blocking type I IFN (IFN-I) signaling through inhibition of STAT2 phosphorylation, which consequently blocks nuclear translocation of STAT1. , SARS-CoV-2 ORF7a also antagonizes restriction of viral replication by bone marrow stromal antigen (BST-2), which acts as a potent inhibitor of viral egress.
ORF7b
The ORF7b protein in SARS-CoV-2 is 43 amino acids long, with approximately 60% sequence similarity to SARS-CoV. The protein contains a transmembrane domain with a putative leucine zipper that may promote multimerization. SARS-CoV-2 ORF7b has been shown to play a role in the attenuation of IFN-I signaling by suppressing STAT1/STAT2 phosphorylation, a step that is essential for their functional activation.
ORF8
ORF8 encodes a 121–amino acid accessory protein displaying less than 20% sequence identity to its counterpart in SARS-CoV. ORF8 contains a predicted Ig domain. The structure of SARS-CoV-2 ORF8 as revealed by x-ray crystallography describes a core that is similar to ORF7a, with the additional presence of two dimerization interfaces. ORF8 is likely to be a secreted protein because ORF8 antibodies represent one of the major markers of SARS-CoV-2 infection. , ORF8 plays a putative role in modulating the host antiviral immune response and has been reported to downregulate major histocompatibility complex (MHC) expression. ORF8 protein also directly interacts with the interleukin-17 (IL-17) receptor A (IL17RA) and activates signaling through the IL-17 pathway, leading to nuclear factor-kappa B (NF-κB) activation and proinflammatory cytokine secretion. ,
ORF9b
ORF9b is an alternative ORF within the nucleocapsid (N) gene. In infected cells, SARS-CoV-2 RNA activates IFN signaling through the RIG-I-MAVS–dependent pathway. Like its ortholog in SARS-CoV, ORF9b protein localizes to the mitochondrial membrane and suppresses IFN-I signaling by blocking MAVS activation. , ORF9b additionally blocks IFN production by inhibiting NF-κB activation.
ORF10
As the final ORF located at the 3′ end of the SARS-CoV-2 genome, ORF10 may code for a putative protein of 38 amino acids containing an alpha-helical region. It does not share sequence similarity with known proteins from SARS-CoV. Through exogenous expression of ORF10, a study reported that the protein could interact with members of a cullin-2 RING E3 ligase complex, specifically with ZYG11B, leading to the hypothesis that ORF10 may be able to hijack the function of ubiquitin ligase complexes. Another study confirmed the interaction with ZYG11B, but unearthed no evidence to indicate that ORF10 could regulate the function of the E3 ligase complex or that this interaction may have any impact on viral processes. The annotation of ORF10 as an ORF in the SARS-CoV-2 genome has been called into question by studies noting the challenge of detecting subgenomic reads for its transcript, indicating that such a protein may not be produced by the virus.
Nonstructural Proteins
ORF1ab
The original Wuhan publication lists ORF1a and ORF1b as separate genes, whereas the NCBI reference sequence (NC_045512.2) combines them under ORF1ab. , Located near the 5′ terminus, ORF1ab spans two-thirds of the genome and encodes the overlapping polyproteins pp1a and pp1ab. The pp1ab polyprotein is generated through a programmed ribosomal frameshifting event. Proteolytic cleavage of the polyproteins by its gene products, the nonstructural protein 5 (Nsp5) and Nsp3 proteases, yields 11 and 15 nonstructural proteins from pp1a and pp1ab respectively (see Fig. 2.1 ). Nonstructural proteins play a variety of roles in the viral replication and transcription complex (RTC), including RNA synthesis, processing, and proofreading ( Table 2.1 ).
| Protein | Function |
|---|---|
| Nsp1 (virulence factor) | Nsp1 suppresses host gene expression by blocking cellular mRNA nuclear export and shutting down translation of mRNA. |
| Nsp2 | In SARS-CoV, Nsp2 interacts with host cell proteins such as prohibitin 1 and prohibitin 2 and may be involved in the disruption of host cellular processes. , |
| Nsp3 (papain-like cysteine protease, PL pro ) | Nsp3, the largest Nsp, is a transmembrane protein containing multiple functional domains including a Mac1 domain with mono-ADP-ribosyl hydrolase activity. , Through its protease activity, Nsp3 cleaves sites in the pp1a and pp1ab polyproteins to yield Nsps. Nsp3 can also remove ubiquitin (Ub) and the Ub-like protein modifier interferon stimulated gene 15 (ISG15) from cellular proteins. SARS-CoV-2 Nsp3 has a preference for mono-Ub and ISG15 over K48-linked and K63-liked polyUb. , SARS-CoV Nsp3 plays an important role in viral RNA replication by recruiting N protein to the RTC and promotes formation of double-membrane vesicles that house viral replication complexes. |
| Nsp4 | Nsp4 is a transmembrane glycoprotein. Interactions of Nsp4 with Nsp3 and Nsp6 drive the formation of double-membrane vesicles for viral replication. |
| Nsp5 (3C-like protease, 3CL pro ; main protease, M pro ) | Nsp5, the viral chymotrypsin-like cysteine protease enzyme or main protease, performs proteolytic processing of most of the cleavage sites in the pp1a and pp1ab polyproteins to generate the component Nsps. Because of its critical role in the generation of viral Nsp proteins, Nsp5 is indispensable for virion production. |
| Nsp6 | Nsp6 is a transmembrane protein that is involved in double-membrane vesicle formation and interacts with Nsp4. It also plays a role in autophagosome formation and maturation. , |
| Nsp7 | Nsp7 forms the primase complex together with Nsp8, and acts as an accessory subunit of the RNA-dependent RNA polymerase complex. The primase complex performs de novo initiation and primer extension. Nsp7 plays a crucial role in RNA binding by the Nsp7-Nsp8-Nsp12 complex. , |
| Nsp8 | Nsp8 possesses RNA-dependent RNA polymerase activity. Nsp8 demonstrates de novo initiation and primer extension activity during RNA synthesis, and forms a complex with Nsp7. Primase activity lies in the N-terminal of Nsp8 and requires the formation of large oligomeric complexes to bring the active site residues into proximity. |
| Nsp9 | Nsp9 is a dimeric RNA-binding protein with a preference for single-stranded nucleic acids. Nsp9 is essential for replication. , |
| Nsp10 | Nsp10 is an essential cofactor for Nsp14 and Nsp16, forming a part of the exonuclease and RNA-capping subcomplex. Nsp10 can bind to single- and double-stranded RNA and DNA. , |
| Nsp11 | Nsp11 is a short peptide with unknown function that forms the final Nsp in the pp1a polyprotein. In pp1ab, the N-terminal sequence of Nsp11 lies between the Nsp10/11 junction and the ORF1ab frameshift site, and becomes the N-terminal part of Nsp12. |
| Nsp12 (RdRp) | Nsp12 is the main RNA-dependent RNA polymerase (RdRp) that performs viral RNA synthesis. During viral RNA replication, RdRp forms a complex with Nsp7, Nsp8, and proteins involved in proofreading and capping. |
| Nsp13 | Nsp13 is a helicase and RNA-5′-triphosphatase. Nsp13 helicase activity is stimulated by binding to Nsp12. Nsp13 unwinds double-stranded DNA and RNA in a 5′–3′ direction, a function that is involved in genomic RNA synthesis by the polymerase complex. Additionally, Nsp13 may facilitate proofreading by stimulating backtracking by the viral replication and transcription complex. |
| Nsp14 | Nsp14 is a bifunctional enzyme with 3′–5′ exoribonuclease activity and N7-guanine methyltransferase activities involved in maintaining replication fidelity and performing 5′-RNA capping respectively. It forms a part of the exonuclease and capping subcomplex, which associates with the Nsp7-Nsp8-Nsp12 polymerase complex. |
| Nsp15 | Nsp15 is a uridine-specific endoribonuclease that preferentially cleaves RNA on the 3′ side of uridines. Nsp15 may be involved in immune evasion by viral RNA processing. Polyuridine tracts at the 5′-end of negative-strand viral RNA intermediates can provoke an IFN response. Through its nuclease activity, Nsp15 may prevent activation of host antiviral sensors by regulating the length of the polyuridine tails and cleaving other sites within the viral RNA to limit the formation of double-stranded RNA (dsRNA) intermediates. |
| Nsp16 | Nsp16 is a 2′-O-methyltransferase that functions as an integral component of the RNA capping machinery. Nsp16 methylates the 5′ cap of viral RNA to mimic human mRNA and prevent viral recognition by the sensor MDA5 in the host cell. Nsp16 was shown to be indispensable for coronavirus replication in cell culture. |
a The functions of nonstructural proteins Nsp1-16 are summarized. This information is based on recently published research about their functions in SARS-CoV-2 or based on knowledge about the functions of their SARS-CoV counterparts. ADP, Adenosine diphosphate; IFN, interferon; MDA5, melanoma differentiation–associated 5; mRNA, messenger ribonucleic acid; Nsp, nonstructural proteins; RTC, replication and transmission complex.
Sequencing and Variants
Genome sequences of SARS-CoV-2 strains detected around the world have been deposited in the GISAID repository, providing a valuable resource for tracking temporal and geographic variations in sequence over the course of the pandemic. The first SARS-CoV-2 sequences derived from patients in Wuhan were almost identical to each other, indicating a recent and common origin for the virus. , , ,
In general, RNA viruses have higher mutation rates compared with DNA viruses, because RNA-dependent RNA polymerases typically lack proofreading mechanisms that limit the incorporation of mutations into the viral genome. However, coronaviruses and a few other related RNA viruses of the order Nidovirales represent an exception and demonstrate lower mutation rates compared with other RNA viruses. For reference, the SARS-CoV-2 genome has been reported to accumulate mutations at a rate that is half the mutation rate of influenza and one-quarter the mutation rate of human immunodeficiency virus. This difference may be attributed to the function of the Nsp14 exoribonuclease in SARS-CoV-2, a proofreading enzyme that is essential for the maintenance of viral genome integrity in coronaviruses. Although mutations in the SARS-CoV-2 genome are expected to occur, most mutations are likely to be neutral or mildly deleterious, and only a minority are likely to confer any fitness advantage to the virus. Nevertheless, monitoring sequence variations in the viral genome, with a focus on mutations in proteins that may have an impact on the behavior of the virus and affect infectivity, transmissibility, pathogenicity, and antigenicity, is integral to the effort of containing the pandemic.
Deletions involving ORF7a, ORF7b, and ORF8 constituted the early emergent variants, detected in the January to February 2020 time frame. , The most common variant was a deletion of 382 nucleotides that truncated ORF7b and deleted most of ORF8, including the transcription regulatory sequence. A 29-nucleotide deletion in ORF8 that was associated with reduced virulence had previously been detected in SARS-CoV strains at the mid-to-late phase of the SARS epidemic. However, the impact of such deletions on infectivity and pathogenicity could not be evaluated because the SARS epidemic came to a natural end. Evaluation of the 382-nucleotide deletion variant in SARS-CoV-2 indicated that the virus retained its replicative fitness, but the deletion may have altered the immune-evasive function of ORF8, resulting in an enhanced immune response to the virus. This variant was linked to milder disease severity and a reduction in the systemic release of proinflammatory cytokines. Consistent with these observations, the 382-nucleotide deletion variant was not detected in patient samples beyond March 2020.
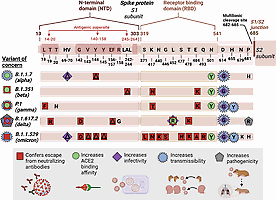
Mutations emerging in the S1 subunit of the spike protein are of special interest because of the critical role of this domain in host cell receptor binding and recognition by neutralizing antibodies ( Fig. 2.4 ). The NTD is targeted by antibodies that specifically recognize epitopes in this region; epitope binning of a large number of NTD-specific monoclonal antibodies (mAbs) revealed multiple antigenic sites. However, one particular site that included residues 14-20, 140-158, and 245-264 was recognized by all known NTD-specific antibodies and aptly named an “NTD supersite.” Beyond the NTD, the RBD in the S1 subunit is immunodominant and represents the primary target of neutralizing antibodies. Therefore mutations in this region can contribute to immune escape. However, mutations emerging in the RBD must not be significantly damaging to hACE2 binding and virus entry into host cells. Outside of the RBD and the NTD, other mutations in the S1 subunit of spike protein may also impact SARS-CoV-2 infectivity and transmissibility.
In March 2020, a variant strain of SARS-CoV-2 that harbored a D614G (23403A>G) substitution in the S1 subunit of the spike protein spread rapidly to become the predominant strain worldwide. By June 2020, this strain, represented by clade “G” (also called clade A2a or type VI), and its offspring “GH” and “GR,” grew to become the most common clades, representing 74% to 78% of all sequenced SARS-CoV-2 genomes. , Studies functionally characterizing the D614G mutation demonstrated increased infectivity of viruses harboring this mutation when tested using both pseudotyped viruses and isogenic recombinant SARS-CoV-2. , Enhanced viral replication was observed in primary human airway epithelial cells, including bronchial and nasal airway epithelial cell cultures. , , This was consistent with reports that the D614G mutation was associated with higher viral loads in the upper respiratory tract of patients. , , There was no indication of increased pathogenicity in animal models, which agreed with the observed lack of association of the D614G variant with altered mortality or clinical severity. , , Hamster and ferret models are useful tools to study SARS-CoV-2 transmission because of their susceptibility to infection and the similarity of the disease developed by the animals to the pan-respiratory moderate to severe COVID-19 and upper respiratory tract–localized mild infection observed in humans, respectively. In hamster and ferret models of SARS-CoV-2 infection, the D614G mutation–harboring virus demonstrated increased transmissibility. , D614G was further shown to enhance viral entry into hACE2–expressing cells. , , At the structural level, D614G disrupted a critical interprotomer contact and shifted S protein conformation toward the “open” or “up” ACE2 binding–competent state to promote membrane fusion with target cells. , , Studies using pseudotyped viruses suggested that D614G could affect S protein processing and shedding or increase functional S protein incorporation into virions. , , However, experiments performed using isogenic viruses expressing additional SARS-CoV-2 structural proteins did not corroborate these effects on S protein processing and incorporation. Viruses that harbored the D614G mutation were as susceptible to neutralization by antibodies and perhaps even slightly more susceptible. , ,
Since the beginning of the pandemic, various SARS-CoV-2 strains have emerged in different parts of the world. As of February 2022, the World Health Organization (WHO) has categorized five strains as variants of concern—strains B.1.1.7, B.1351, P.1, B.1.617.2, and B.1.1.529.
The B.1.1.7 variant (also known as 501Y.V1; WHO label: alpha) that first emerged in the United Kingdom harbors 17 nonsynonymous mutations, including the D614G mutation and 8 additional mutations in the spike protein: ΔH69-V70, ΔY144, N501Y, A570D, P681H, T716I, S982A, and D1118H. Preliminary evidence suggests that this variant may possess increased transmissibility and may be associated with an increased risk for mortality. The only mutation localized to the RBD, N501Y, has been reported to increase hACE2 binding affinity, which likely signifies an increase in the ACE2 binding affinity of the B.1.1.7 variant over the original strain. , Although mice normally demonstrate poor susceptibility to SARS-CoV-2 infection because of suboptimal recognition of the mouse ACE2 receptor, the N501Y mutation facilitates adaptation and successful infection by SARS-CoV-2 in a mouse model; therefore N501Y may enhance cross-species transmission. , However, there is no evidence to indicate that the N501Y mutation increases infectivity of the virus in the context of hACE2–expressing cells. The P681H mutation is located adjacent to the furin cleavage site (RRAR) that spans amino acids 682-685. To a limited extent, the P681H mutation enhances spike protein cleavage and fusogenic potential of the alpha variant, although this mutation alone does not increase virion infectivity in vitro. , The ΔH69-V70 deletion increases infectivity, enhances incorporation of cleaved spike protein into virions, and accelerates syncytium formation. , , The ΔY144 deletion is located within the NTD supersite and confers resistance to NTD-specific monoclonal antibodies. Overall, the B.1.1.7 variant is less sensitive to some monoclonal antibodies, but in general remains susceptible to neutralizing antibodies. , The combination of spike mutations in this variant may be associated with a modest, if any, reduction in vaccine efficacy.
B.1351 is a variant of concern that also harbors the N501Y mutation found in the B.1.1.7 strain. The B.1351 variant (also known as 501Y.V2; WHO label: beta), which was first detected in South Africa, harbors eight lineage-defining mutations in the spike protein, including the D614G mutation: D80A, D215G, ΔL242-L244, K417N, E484K, N501Y, and A701V. This variant shows no apparent difference in infectivity but greatly enhances immune escape by conferring resistance to neutralization by mAbs and convalescent and vaccine-elicited polyclonal sera; this observation has raised much concern in the international community. , Additionally, because of the combination of mutations in the RBD, the B.1351 variant is reported to possess increased ACE2 binding affinity, higher than that of B.1.1.7. Multiple studies have shown that the E484K mutation confers resistance to neutralizing antibodies. , In addition, deep mutational scanning in yeast indicates that E484K might slightly increase ACE2 binding affinity. Mutations at K417, including K417N and K417T, occur in multiple variants of interest, and have been identified as escape mutations that can cause resistance to monoclonal neutralizing antibodies. , , Beyond the established effects of the RBD mutations E484K, K417N, and N501Y, the deletion of residues 243-244 also has been shown to cause resistance to NTD-specific neutralizing antibodies.
The P.1 variant (also known as 501Y.V3; WHO label: gamma) which was first detected in Brazil, includes 10 new nonsynonymous mutations in the spike protein besides the D614G mutation: L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, H655Y, and T1027I. The P.1 strain shares the triplet combination of mutations found in the B.1.351 variant—K417X, E484K, and N501Y—indicating convergent molecular adaptation. The H655Y mutation has been shown to enhance transmissibility in a hamster model of SARS-CoV-2 infection. The L18F mutation in the antigenic supersite reduces neutralization by NTD mAbs. Overall, the P.1 variant is estimated to possess higher transmissibility and demonstrates an increase in ACE2 binding affinity to an extent similar to that of B.1351. As a result of the presence of nearly identical mutations in the RBD, the P.1 strain also demonstrates a significant degree of escape from mAbs targeting the RBD, in a manner similar to that of B.1351. However, unlike B.1351, the P.1 strain does not show a substantial reduction in neutralization by polyclonal sera, suggesting that it is unlikely to affect vaccine efficacy.
The B.1.617.2 variant (WHO label: delta) that was first detected in India is reported to contain multiple conserved changes in the spike protein, including the D614G mutation: T19R, T95I, G142D, ΔE156-F157, R158G, L452R, T478K, P681R, and D950N. The P681R mutation has been linked to increased spike protein cleavage, enhanced spike protein fusogenicity and syncytium formation, and increased pathogenicity of the delta variant. , Analysis of sequences deposited in GISAID revealed that L452R emerged independently in various strains in the December 2020 to February 2021 time frame. The L452R mutation increases ACE2 binding affinity, enhances entry into cells coexpressing ACE2 and TMPRSS2, increases pseudovirus infectivity against human airway lung organoids, and may increase viral replication. , The L452R mutation additionally confers escape from neutralization by mAbs and polyclonal human immune sera, and evades antigen recognition by human leukocyte antigen (HLA)–restricted cellular immunity. , , The T470-T478 loop is a critical determinant of spike RBD recognition by the ACE2 receptor. The T478K mutation is yet to be extensively characterized, but was found to be enriched upon exposure to weak neutralizing antibodies. Another substitution mutation at the same position, T478I, confers resistance to neutralizing mAbs and polyclonal human immune sera, and the T478S mutation has been demonstrated to increase ACE2 binding. , The ΔE156-F157 and G142D mutations are located within the same NTD supersite as the ΔY144 mutation and may evade recognition by NTD-specific antibodies. In August 2021, B.1.617.2 strain became the dominant strain in the United States and in much of the world. This strain has been reported to possess increased transmissibility and establish much higher viral loads in patients, hinting at enhanced replicative potential and infectivity. , Additionally, B.1617.2 can escape neutralization by antibodies that target both RBD and non-RBD epitopes. Although the existing vaccines remain effective against B.1.617.2, their efficacy may be reduced. ,
The B.1.1.529 variant (WHO label: omicron) was identified in Botswana and South Africa in November 2021. Within a few weeks, the omicron variant spread rapidly across the globe and became the predominant source of new Covid-19 cases. B.1.1.529 contains over 30 non-synonymous mutations in spike protein. It shares many mutations in spike protein with other variants of concern, including – ΔH69-70, T95I, G142D, K417N, T478K, N501Y, D614G, H655Y, and P681H; additionally, the E484A and Δ143-145 mutations are similar to previously observed mutations that confer escape from neutralizing antibodies. , B.1.1.529 also contains an insertion mutation (ins214EPE), a small deletion mutation (ΔN211), and several novel mutations – A67V, L212I, G339D, S371L, S373P, S375F, N440K, G446S, S477N, Q493R, G496S, Q498R, Y505H, T547K, N679K, N764K, D796Y, N856K, Q954H, N969K, and L981F. Many of the mutations affecting the RBD and the NTD are expected to cause resistance to neutralizing antibodies. , , , The omicron variant escapes neutralization by convalescent sera from patients previously infected with other SARS-CoV-2 variants and with the two-dose series of mRNA vaccines; however, booster doses are reported to reinstate protection against this variant. – , , Various studies have reported that the RBD of the omicron variant either possesses increased or similar ACE2 binding affinity compared to the original Wuhan strain. , , , Preliminary evidence suggests that the omicron variant is highly transmissible. Unlike the delta variant, the omicron variant does not efficiently infect the lungs but replicates successfully in the bronchus and in the upper respiratory tract. , This may be linked to the route of host cell entry employed by the omicron variant; preliminary studies indicate that omicron uses the cathepsin-mediated endosomal route of cell entry instead of TMPRSS2-dependent membrane fusion. , Despite the presence of mutations such as P681H and H655Y which have been shown to increase S protein cleavage, the omicron variant displays reduced fusogenic potential and decreased syncytia formation compared to other variants. , In addition to its inability to effectively infect lung cells, this may contribute to the attenuation in pathogenicity reported for the omicron variant. However, further investigations are necessary to fully characterize the omicron variant. While much research has been performed to evaluate the effect of individual mutations observed in spike protein, it is important to keep in mind that the properties of a SARS-CoV-2 variant are dictated by the sum total of mutational effects on Spike protein, and additionally by mutations in other structural, accessory, and nonstructural proteins that have not yet been characterized. To end the pandemic, it is essential to continue monitoring the emergence of new variants and investigate their impact on receptor binding, host cell entry, infectivity, transmissibility, pathogenicity, escape from neutralizing antibodies, and detection by diagnostic tests.
Mechanisms of SARS-CoV-2 Pathogenesis
Receptor Binding and Entry Into Host Cells
Early studies characterizing the SARS-CoV-2 virus revealed that ACE2 is the host cell receptor exploited by the virus for cell entry. , ACE2 is an integral transmembrane protein that functions as a carboxypeptidase and plays an important role in the regulation of blood pressure. The spike protein, which binds to the peptidase domain of ACE2, contains two sites that have to be cleaved to unleash the full infectivity of the virus, a process referred to as S protein “priming.” The S1/S2 junction of SARS-CoV-2 contains a furin cleavage site, which is the first site to be cleaved by host cell proteases. Spike protein cleavage may be carried out in producer cells by furins or furin-like enzymes during trafficking in the secretory pathway or by cathepsins in the late endosome or endolysosome of target cells. Alternatively, cleavage may occur at the target cell surface before viral entry, by transmembrane serine proteases such as TMPRSS2. After S1/S2 cleavage, a second cleavage site becomes exposed, the S2′ site. Cleavage at this site is necessary for liberating the S2 fusion peptide and initiating viral membrane fusion with the host cell membrane.
The furin cleavage site is reported to be an important determinant of viral transmission in ferret models of SARS-CoV-2 infection. Using lentiviral pseudotypes and a cell culture–adapted SARS-CoV-2 virus with an S1/S2 deletion, the polybasic furin cleavage site has been shown to confer a selective advantage to SARS-CoV-2 in lung cells and primary human airway epithelial cells. This selective advantage depends on the expression of the cell surface protease TMPRSS2, which has been demonstrated to be essential for SARS-CoV-2 S protein priming and viral entry into lung cells. , ,
The presence of the polybasic cleavage site in the spike protein provides a unique advantage to SARS-CoV-2 by increasing susceptibility to furin-mediated cleavage in the producer cells. The resulting virions with pre-cleaved spike proteins demonstrate enhanced entry into TMPRSS2-expressing cells in the human airway and lungs. Furin-dependent cleavage of the polybasic site potentiates the infectivity of the virions by promoting the processing of spike in the producer cell and making the S2′ cleavage site accessible to TMPRSS2 at the receiver cell surface. TMPRSS2 cleaves the spike protein at S2′ and promotes early entry at or in the vicinity of the cell surface, rather than late entry through the endosome, which is characteristic of cathepsin-dependent cleavage. By reducing reliance on low pH–dependent and cathepsin-mediated cleavage in the late endosomes, the polybasic furin cleavage site confers escape from restriction by innate immune antiviral proteins belonging to the IFN-induced transmembrane protein family (IFITM), which localize to the late endosomes and prevent the entry of double-enveloped viruses. , Alteration of the furin cleavage site ablates processing by furin and promotes the incorporation of uncleaved spike protein into virions. , Loss of this site is associated with a reduced replication rate and attenuated infection in both hamster and K18-hACE2 transgenic mouse models of SARS-CoV-2 pathogenesis. , Additionally, mutation of the SARS-CoV-2 furin cleavage site eliminates cell–cell fusion, which is a mechanism by which viruses can spread from infected cells to neighboring cells. This is mediated by spike protein from newly formed virions which are released to the cell surface. At the surface, spike protein binds ACE2 to promote fusion with neighboring cells, a phenomenon that is characterized by the formation of large multinucleate cells or syncytia. TMPRSS2 enhances virion infectivity by increasing syncytia formation. Unlike the effect observed with loss of the cleavage site, loss of furin alone does not eliminate cell–cell fusion or infectivity if TMPRSS2 is expressed by the acceptor cell. Therefore, although the polybasic cleavage site is essential for both infectivity and fusogenic activity, cleavage at this site is not exclusively mediated by furin.
The proprotein convertase furin is a type I transmembrane protein that is widely expressed in various cells and tissues. Furin is ubiquitously expressed; thus the expression levels and distribution of ACE2 and TMPRSS2 across various tissues and organs is the factor that likely dictates SARS-CoV-2 organotropism. In general, ACE2 is expressed at lower levels compared with TMPRSS2, but ACE2 and TMPRSS2 are detected in both the nasal and bronchial epithelium. ACE2 is expressed in multiple epithelial cell types throughout the airway, including alveolar epithelial type II (AT2) cells, which play a central role in SARS-CoV-2 pathogenesis. , TMPRSS2 presents a broader distribution and higher expression across various tissue types, suggesting that ACE2 expression may be the limiting factor for initial SARS-CoV-2 infection and tropism. TMPRSS2 is expressed only in a subset of cells expressing ACE2, indicating that the virus may use cathepsins or other proteases for S protein priming in certain cell types. ACE2+, TMPRSS2+ cells in the nasal and airway passages are mostly secretory goblet and multiciliated cells, whereas those in the lungs are AT2 cells. ,
Replication and Assembly
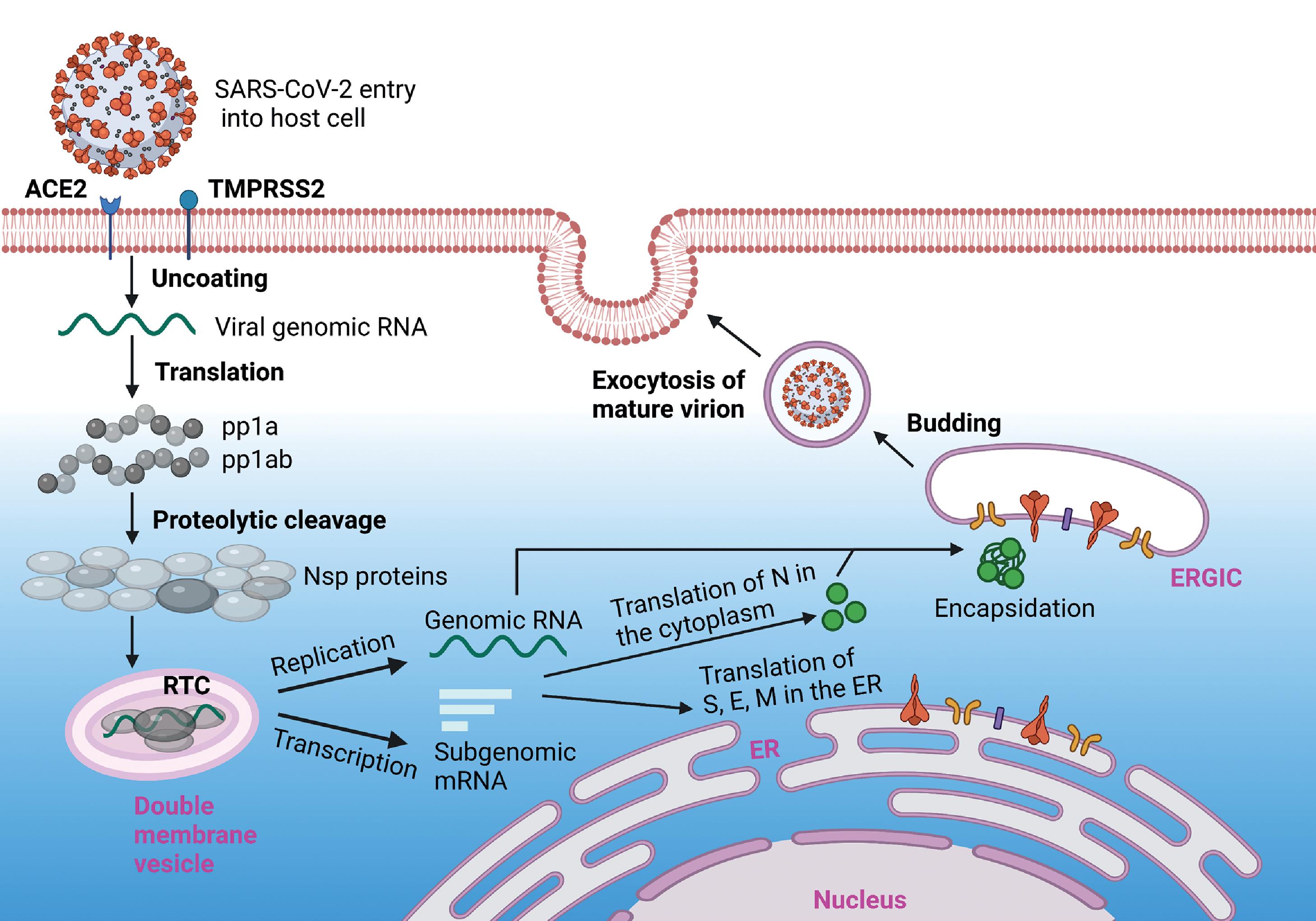
Upon membrane fusion or endocytic entry, viral genomic RNA is released by uncoating and translated by the host cell protein translation machinery. The polyproteins pp1a and pp1ab are proteolytically cleaved by their own component proteins Nsp3 (PL pro ) and Nsp5 (3CL pro , main protease) to generate the full series of viral nonstructural proteins (Nsp1-16). Most of the Nsps are involved in the formation of the viral RTC and function to create a cellular environment that is conducive for viral RNA synthesis, virion assembly, and infection.
Positive-sense viral genomic RNA is used as a template to generate full-length negative-sense copies for replication, and subgenomic negative-sense RNAs to produce the nested set of subgenomic mRNAs (sgmRNAs). , The cytosol of coronavirus-infected cells, such as those infected with SARS-CoV-2, contain a high density of double-membrane vesicles derived from the ER. These double-membrane vesicles constitute the sites of viral genomic RNA replication and subgenomic mRNA transcription, providing a protective and resource-rich environment away from disruptive host cell factors, with dsRNA replication intermediates segregated in the interior. , The viral nonstructural proteins Nsp3, Nsp4, and Nsp6 are implicated in the formation of double-membrane vesicles and in anchoring of the RTC to the vesicular membrane. The RTC includes multiple viral nonstructural proteins, with the core polymerase complex formed by Nsp12, Nsp7, and Nsp8.
Nsp12, the main RdRp, possesses low RNA polymerization activity on its own. However, the primase complex proteins Nsp7 and Nsp8 greatly stimulate its catalytic activity by promoting RNA binding. The SARS-CoV-2 core polymerase complex is formed by the Nsp12 subunit bound to an Nsp7-Nsp8 heterodimer, with an additional Nsp8 subunit bound at a different site. Nsp12 extensively interacts with RNA through the phosphate-ribose backbone, implying sequence-independent binding. The holoenzyme for viral replication includes other Nsp proteins to incorporate proofreading and capping functions, such as the Nsp13 RNA 5′-triphosphatase with helicase activity, and the Nsp10-Nsp14-Nsp16 exonuclease and capping subcomplex.
Double-membrane vesicles housing the RTC contain membrane-spanning molecular complexes formed by Nsp3 that connect the interior of the vesicle with the cytosol. These pores are expected to act as a conduit for the exit of viral sgmRNA and genomic RNA products to sites of translation and encapsidation by N protein, respectively. N protein is translated in the cytoplasm and encapsidates nascent genomic RNA to form the nucleocapsid. Viral structural proteins (S, E, and M) are translated in the ER and transported to the ERGIC, the main site of coronavirus assembly. Interaction with encapsidated genomic RNA in the ERGIC induces budding into the lumen of secretory vesicles, which ultimately fuse with the plasma membrane, releasing the virions from the infected cell by exocytosis. The viral replication process is illustrated in Fig. 2.5 .
Disruption of Host Cellular Processes and Defenses
Coronaviruses hijack the host cell machinery and resources to produce viral proteins and replicate. To do this, CoVs must first block protein synthesis of host cellular mRNA.
Nsp1, the “host shutoff factor,” which plays a multifaceted role in the suppression of host protein synthesis by coronaviruses, undergoes rapid proteolytic release from the polyprotein. SARS-CoV-2 Nsp1 inhibits the nuclear export of cellular mRNAs by interacting with the host mRNA export receptor heterodimer NXF1-NXT1. Through its C-terminal, Nsp1 binds to the 40S ribosomal subunit and occludes the mRNA entry tunnel, thereby suppressing host cellular mRNA translation. Beyond Nsp1, Nsp8 and Nsp9 bind to the signal recognition particle to disrupt host cell protein trafficking, whereas Nsp16 binds to the mRNA recognition domains of U1 and U2 splicing RNAs and affects mRNA splicing at a global level. By disrupting the production of host proteins, including those involved in IFN-I signaling, these nonstructural proteins blunt the host cell innate immune response and antiviral defense mechanisms.
IFN-I and IFN-III induction upon SARS-CoV-2 infection plays a critical role in the innate defense mechanism by limiting viral replication. Type I IFNs (IFN-α and IFN-β) are widely expressed, whereas type III IFN (IFN-λ) responses are mainly restricted to mucosal surfaces. SARS-CoV-2 viral replication has been reported to induce a delayed IFN response in lung epithelial cells. Pathogen-associated molecular patterns (PAMPs), including dsRNA intermediates produced during viral replication, can be recognized by pattern recognition receptors (PRRs) such as retinoic acid–inducible gene I (RIG-I), melanoma differentiation–associated 5 (MDA5), and Toll-like receptor 3 (TLR3). In SARS-CoV-2–infected lung epithelial cells, MDA5 and laboratory of genetics and physiology 2 (LGP2) protein were primarily involved in the IFN response, with MDA5 acting as the main viral dsRNA sensor. MDA5 triggers formation of the mitochondrial antiviral-signaling protein (MAVS) signaling complex consisting of proteins MAVS-TRAF3-TRAF6-TOM70 on the mitochondrial outer membrane. Signaling by this complex leads to the activation of TANK-binding kinase 1 (TBK1), which phosphorylates IFN regulatory factor 3 (IRF3), promoting its nuclear localization and subsequent transcriptional activation of IFN-β. IRF3, IRF5, and NF-κB drive IFN-β and IFN-λ induction in lung epithelial cells infected with SARS-CoV-2. Secreted IFN-β and IFN-λ bind to and activate their respective receptors on infected cells and neighboring cells, which leads to phosphorylation and heterodimerization of STAT1-STAT2. The STAT1-STAT2 heterodimer recruits IRF9 to form ISGF3, which then translocates to the nucleus. ISGF3 binds to IFN-stimulated response elements (ISRE) in the promoters of hundreds of genes with antiviral functions, called IFN-stimulated genes (ISGs)=, and induces their expression. Proteins encoded by ISGs not only induce strong antiviral innate immune responses but also include MHC molecules, which play a critical role in regulating acquired immunity.
The type II interferon, IFN-γ, signals through a distinct pathway in which binding of the IFN to its receptor complex induces STAT1 phosphorylation and homodimerization to produce the IFN-g activated factor (GAF). GAF translocates to the nucleus and activates genes that contain gamma-activated sequence (GAS) promoter elements. Through these different types of IFNs, STAT1 can activate different sets of ISGs. Importantly, the genes activated by the STAT1-STAT2 heterodimer regulate the innate immune response on viral RNA sensing, whereas the genes activated by the STAT1 homodimer induce proinflammatory responses and promote macrophage activation.
Coronaviruses have evolved complex mechanisms to escape viral recognition and suppress the IFN response. In addition to forming double-membrane vesicles to shield dsRNA intermediates that can be recognized by cytosolic PRRs, viral mRNA is “capped” by Nsp14 and Nsp16 and modified by Nsp15 to prevent recognition by PRRs. Capping of viral RNA at the 5′ end consists of an N-methylated guanosine triphosphate and a C2′-O-methyl-ribosyladenine and serves to disguise viral RNA to resemble human mRNA. It also ensures efficient translation by the cellular machinery and stabilizes the RNA.
The de-ISGylation and de–adenosine diphosphate (ADP)–ribosylation activities of Nsp3 play a role in the evasion of innate immunity. ISGylation is a posttranslational modification similar to ubiquitination in which ISG15, a small protein that is highly induced by IFN-I, is conjugated to target proteins and modulates their functions. Interestingly, SARS-CoV-2 Nsp3 preferentially cleaves ISG15, whereas its SARS-CoV counterpart preferentially cleaves polyubiquitin chains. MDA5-mediated viral RNA sensing depends on the ISGylation of its caspase activation and recruitment domain (CARD), which promotes its oligomerization. By actively performing de-ISGylation, Nsp3 antagonizes MDA5 activation. Additionally, Nsp3 cleaves ISG15 from IRF3, which leads to decreased phosphorylation and reduced nuclear translocation of IRF3. By mimicking host cell sensing of viral nucleic acids through treatment with poly(I:C) which normally induces IFN-β expression, expression of SARS-CoV-2 Nsp3 was observed to decrease the activation of the IFNB1 promoter more effectively compared with SARS-CoV Nsp3. SARS-CoV-2 Nsp3 also contains a Mac1 domain, which can remove mono-ADP ribose modifications generated by PARP14 on host cell protein substrates. , These modifications are involved in the IFN response, and their removal alters STAT1 regulation and hinders IFN production. Mutation of the Mac1 domain in SARS-CoV Nsp3 induced IFN production and resulted in attenuated infection in a mouse model. Therefore, through these different mechanisms, SARS-CoV-2 Nsp3 is expected to play a key role in attenuating the IFN-I response.
Several other SARS-CoV-2 proteins also antagonize IFN signaling at various levels by inhibiting IRF3 nuclear translocation, by blocking STAT1/STAT2 phosphorylation and nuclear translocation and by suppressing ISG transcription ( Fig. 2.6 ). Nsp13 binds to and blocks TBK1 phosphorylation, and Nsp6 binds to TBK1 and inhibits IRF3 phosphorylation. ORF6 inhibits both STAT1 and IRF3 nuclear translocation, whereas ORF3b, Nsp14, and Nsp15 block IRF3 nuclear translocation. , , Nsp6, Nsp13, and ORF7b suppress STAT1/STAT2 phosphorylation. Nsp1, ORF3a, and M protein inhibit STAT1 phosphorylation, whereas ORF7a inhibits STAT2 phosphorylation. SARS-CoV-2 N protein can suppress IFN-b induction by two mechanisms: by interacting directly with RIG-I through its DExD/H-box helicase domain, which is responsible for viral RNA sensing, and by binding and inhibiting STAT1/STAT2 phosphorylation and nuclear translocation. , ORF9b protein localizes to the mitochondrial membrane to suppress IFN-I signaling by associating with the substrate-binding site of TOM70, which is involved in the activation of MAVS. , Additionally, during SARS-CoV-2 infection in primary human pulmonary alveolar epithelial cells, ORF9b has been shown to accumulate and antagonize IFN signaling by interrupting the K63-linked polyubiquitination of NF-κB essential modulator (NEMO) protein, consequently inhibiting canonical IKKα/β-NEMO-NF-κB signaling and blocking IFN production. SARS-CoV-2 M protein functions as an IFN antagonist by directly interacting with MAVS and preventing the formation of the RIG-I/MDA5-MAVS-TRAF3-TBK1 signaling complexes. ,