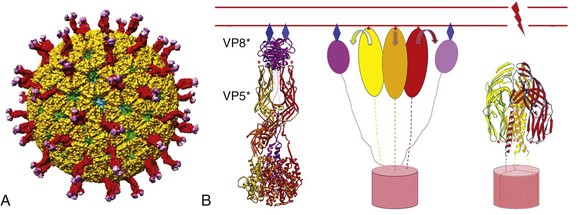
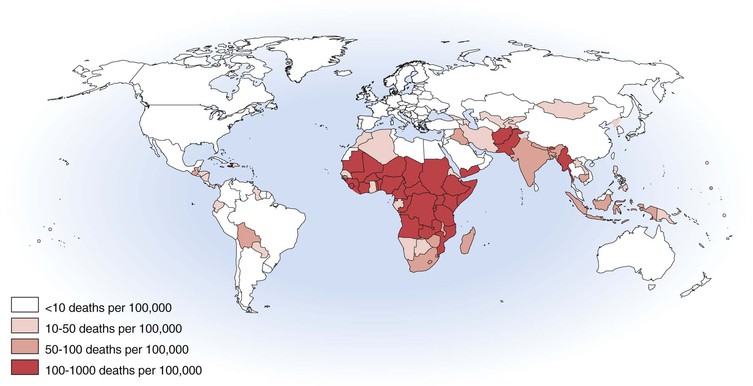
Philip R. Dormitzer Rotavirus is a genus within the Reoviridae, a family of nonenveloped, icosahedral animal viruses with double-stranded, segmented RNA genomes. The family Reoviridae also includes reoviruses, which have occasionally been isolated from patients with respiratory illnesses but have not been established as a cause of human disease, and coltiviruses and seadornaviruses, which cause disease in humans (see Chapters 150 and 151). Rotaviruses are the most important cause of severe, dehydrating gastroenteritis in children younger than age 5 years in all socioeconomic groups and in all regions of the world. As of 2008, they were responsible for approximately 450,000 infant and childhood deaths annually, with more than half of those deaths occurring in India, Nigeria, Pakistan, the Democratic Republic of Congo, Ethiopia, and Afghanistan.1 Higher latitude and higher income correlate with a seasonal pattern of disease in cold and dry months; lower latitude and income correlate with less distinct seasonality, although neither geography nor income fully account for observed annual patterns.2 Before the association of rotaviruses with human disease, etiologic agents were not identified in most cases of childhood gastroenteritis. In 1973, electron microscopic examination of duodenal biopsy specimens from six of nine children with acute gastroenteritis revealed similar viral particles, which were approximately 70 nm in diameter.3 Morphologically, these particles were indistinguishable from viruses previously identified in specimens from mice and cows with diarrhea4,5 and were designated rotaviruses because of their appearance in electron micrographs as wheels with spokes (Fig. 152-1). The antigenic similarity between the human and bovine agents was confirmed when an exchange of matched liquid stool and convalescent sera between a veterinary and a medical laboratory showed that antibodies in the sera of children and calves agglutinated the rotavirus particles in the stools of both species.6 The human sera also neutralized the infectivity of the bovine agent, which had been adapted to growth in cell culture.6 Since the discovery of the agent, understanding of rotavirus replication, pathogenesis, and immunity has relied on the study of strains that grow well in cell culture and on large and small animal models for rotavirus infection and disease. The ability to “mate” rotavirus strains by coinfecting cells to achieve reassortment of the 11 genome segments has allowed classic genetic studies. Extensive global epidemiology has relied on an elaborate serology, now supplemented by nucleic acid–based diagnostics. This scientific background enabled the introduction of live-attenuated vaccines for human use. These vaccines have greatly reduced the health impact of rotaviruses in the upper and middle income countries where they have been introduced, with evidence of both direct vaccine-elicited immunity and a herd effect.7 Efforts to extend effective immunization to the most severely affected populations in developing countries are underway.8 The application of molecular and structural biology provides the scientific basis for the next generation of vaccines. The rotavirus virion is a nonenveloped icosahedral particle, which is approximately 770 Å in diameter, excluding the VP4 spikes (Fig. 152-2A).9,10 It consists of three concentric protein layers, which encapsidate 11 segments of tightly packed, double-stranded RNA, together with polymerase and capping enzyme complexes. Each genome segment contains one or, in the case of genome segment 11, two open reading frames. These segments encode six structural proteins (VP1 to VP4, VP6, and VP7) and six nonstructural proteins (NSP1 to NSP6). The innermost protein shell consists of 120 copies of VP2, arranged in an icosahedral lattice (T = 1 with two VP2 molecules making up each icosahedral asymmetrical unit).9 The VP1 polymerase and VP3 capping enzyme are anchored inside the VP2 shell adjacent to pores at the icosahedral fivefold vertices. The middle layer consists of 780 copies of VP6, which form thick trimeric pillars in an icosahedral lattice (T = 13 levo). Although not exposed on the virus surface, VP6 is the target of the most abundant antibodies elicited by rotavirus infection. The genome, VP1, VP3, and the inner two protein layers make up the transcriptionally active, double-layered subviral particle (DLP). The thin outermost layer consists of 780 copies of a coat glycoprotein, VP7, and 180 copies of a spike protein VP4, which forms asymmetrical trimers that protrude from the virion (see Fig. 152-2B, left).10 One hundred and thirty-two aqueous holes perforate the outer and middle layers. VP4 and VP7 translocate the DLP across a host membrane and into the cytoplasm. Both outer layer proteins are neutralization antigens.11 VP4 is the major cell attachment protein, a membrane penetration protein, and a determinant of virulence.11–14 VP7 forms the icosahedral lattice of the outermost layer, which is in register with the VP6 lattice of the middle layer. The rotavirus particle is physically hardy and resists inactivation by treatment with fluorocarbons, ether, and concentrations of chlorine typically used to treat sewage effluent and drinking water.15 However, the particle is inactivated by calcium chelators and by antiseptic agents that contain relatively high concentrations of alcohols (>40%), free chlorine (>20,000 ppm), or iodophors (>10,000 ppm iodine).16 A commonly used hand-sanitizing agent (Purell), which contains 62% ethanol, substantially reduces viable rotavirus carriage on fingertips,17 and a disinfectant spray (Lysol), which contains 79% ethanol and 0.1% o-phenylphenol, prevents the experimental transmission of rotavirus from fomites to human volunteers.18 Rotavirus survival in the environment is significantly decreased at high relative humidity.15 Efficient rotavirus infectivity requires priming of the virus by intestinal trypsin, which cleaves the spike protein, VP4, into an amino-terminal fragment, VP8*, and a carboxy-terminal fragment, VP5* (see Fig. 152-2). The VP8* fragment contains a hemagglutination domain, and the VP5* fragment contains a β-barrel domain that interacts with membranes.10 Cleavage into VP5* and VP8* triggers an initial rearrangement of VP4 trimers into an asymmetrical conformation (see Fig. 152-2B, left) in which the three subunits form a “foot,” buried under the VP7 shell with approximate threefold symmetry, and the β-barrel domain of VP5* of one subunit forms a “stalk” that supports a “body,” formed by the β-barrel domains of VP5* from each of the other two subunits.10 The spike is topped by two “heads,” formed by the central VP8* domains of two of the subunits.10 The rotavirus cell entry pathway is complex, with entry-related conformational changes in VP4 and VP7, strain-dependent variation in use of cell surface receptors, and strain-dependent variation in the use of different endocytic pathways.19,20 The first rotavirus interaction with a cell generally occurs when the VP8* heads bind a cell surface glycan (see Fig. 152-2B). For many rotavirus strains that cause disease in nonhuman animals, the host cell glycan is a neuraminidase-sensitive terminal sialoside on cell membrane glycoproteins or glycolipids; for some strains that cause disease in humans, the initial receptor is a neuraminidase-insensitive nonterminal sialoside or a histo-blood group antigen that does not contain sialic acid.21,22,23 After initial attachment to a carbohydrate, the virus may interact with host protein receptors, such as heat shock protein 70 or one of several integrins (including α2β1, αvβ3, and αxβ2).24 Cell entry requires the action of dynamin and the presence of cholesterol in cellular membranes.20 Some strains enter through clathrin-mediated endocytosis; others through a clathrin- and caveolin-independent endocytic pathway.20,25 Endosomal acidification does not seem to be required for entry, as it is for many other viruses, but falling calcium concentration is probably necessary.25a In low calcium environments, calcium is lost from the VP7 intratrimer interface and the VP7 trimers dissociate, releasing clamplike contacts with VP6 so that VP7 is shed from the virion.26,27 When VP7 uncoating occurs in the presence of membranes, cleaved VP4 rearranges to an inferred intermediate conformation that binds the membranes (see Fig. 152-2B, center).28 This intermediate conformation resolves into a post–membrane-penetration state in which the VP5* fragments form an umbrella-shaped, threefold symmetrical, folded-back trimer (see Fig. 152-2B, right).29 During these conformational changes, the hydrophobic apex of VP5* is translocated from one end of the molecule to the other.29 This motion resembles the fold-back rearrangements of enveloped virus fusion proteins, which mediate fusion between a virus envelope and a cellular membrane. Loss of membrane penetration when the VP5* apices are mutated to reduce their hydrophobicity suggests that the “jackknifing” of VP5* breaches a cellular membrane.14 The membrane breach must be substantial. Because naked rotavirus double-stranded RNA genome segments are not infectious, the entire transcriptionally active 710-Å DLP must be translocated into the cytoplasm to initiate infection. The loss of VP7 leads to conformational shifts deeper in the particle, near the polymerase complex below the channels at the fivefold vertices, to activate the transcriptase activity of the rotavirus DLP so that, once in the cytoplasm, the DLP extrudes capped, nonpolyadenylated messenger RNA (mRNA) through channels at its fivefold icosahedral vertices.27,30,31 The genome remains encapsidated throughout transcription. The translation of viral mRNAs is enhanced by circularization, mediated by the specific binding of the rotavirus nonstructural protein NSP3 to a four-nucleotide consensus sequence at the 3′ end of viral mRNA and to the cellular cap-binding protein eIF4G.32–34 Thus, NSP3 substitutes for a cellular polyadenylic acid binding protein to favor the translation of the nonpolyadenylated viral mRNAs over polyadenylated host mRNAs. Rotavirus genome replication must overcome two major challenges: one copy of each of 11 different RNAs must be selected for packaging, and the newly synthesized double-stranded RNA must be sequestered so that it does not induce an interferon response. The mechanism of selective packaging remains obscure but may involve base-pairing interactions between the nascent genome segments; the mechanism of genome sequestration is better understood.35 The viral polymerase, VP1, specifically binds rotavirus mRNAs by recognizing a consensus sequence (UGUG) near their 3′ ends but holds the mRNAs in an inactive complex.36 The mRNA shifts to a position that permits negative strand synthesis only when the polymerase binds to the inner face of a decamer of the inner shell protein, VP2.37 The dependence of negative-strand synthesis on VP1-mRNA complexes binding to the inner shell protein ensures that double-stranded genomic RNA is synthesized within a nascent particle. Several viral nonstructural proteins are involved in genome replication and the assembly of precursor particles.35 DLPs are assembled in cytoplasmic viral “factories,” termed viroplasms, which form through the interaction of rotavirus nonstructural proteins NSP2 and NSP5.38 NSP2 is an octomeric doughnut-shaped structure with RNA-binding, helix-destabilizing, nucleoside triphosphatase, and nucleoside diphosphate kinase–like activities.39 Activities proposed for NSP2 include acting as a molecular motor to drive replicating mRNA into the nascent particles and maintaining nucleotide pools in viroplasms during replication.39,40 NSP5 is metalloprotein that binds an iron-sulfate cluster and also a phosphoprotein that has autokinase activity.41,42 NSP5 probably has a regulatory role, competing with single-stranded RNA to bind in grooves of the NSP2 octamer, competing with VP6 for binding to VP2, and binding RNA.43,44 Once VP6 is added to the nascent particle, the newly formed DLP can transcribe mRNA, amplifying virus yield. Newly transcribed rotavirus mRNA enters two pools: mRNA that remains sequestered within viroplasms is replicated and packaged as genome segments; mRNA that escapes viroplasms is translated.45 Despite the barrier imposed by the need to introduce recombinant mRNA into viroplasms for replication, recombinant genes have been incorporated into infectious rotavirus genomes.46,47 The reverse genetics system is complicated and involves transfecting cells with a plasmid bearing a T7 promoter-controlled recombinant gene, infecting with a vaccinia virus expressing T7 polymerase, coinfecting with a helper rotavirus, and selecting against the wild-type helper virus progeny by antibody neutralization, temperature sensitivity, or small interfering RNA (siRNA) gene silencing. As reverse genetic rescue becomes more efficient, the technique promises accelerated basic discovery and, potentially, rationally attenuated vaccine strains. After genome replication and DLP assembly, the outer capsid is added through an unusual maturation process. The DLP binds the cytoplasmic tail of NSP4, a rotavirus nonstructural protein that is resident in the membranes of the endoplasmic reticulum (ER) and viroplasm-associated vesicles, and then buds into the ER lumen, transiently acquiring an envelope.48 VP7, also resident in the ER, mediates removal of the transient envelope and binds the DLP, locking the VP4 spike protein in place.49,50 Thus, rotavirus penetrates membranes twice. During entry, rotavirus penetrates a cellular membrane through a mechanism involving rearrangements of cleaved VP4 as the outer capsid disassembles; during maturation, rotavirus penetrates an envelope derived from the ER through a mechanism that involves VP7 and possibly NSP4, as the outer capsid assembles. Rotavirus egress from infected enterocytes is accomplished by release from the apical surface after vesicular transport from the ER by a pathway that bypasses the Golgi apparatus.51 Infants and young children with diarrhea caused by rotavirus are more likely to have severe symptoms and become dehydrated than patients with diarrhea related to other common enteric pathogens.52,53 The clinical manifestations of rotavirus gastroenteritis in humans have been studied in experimental infections in adults. In one such study, 4 of 18 adult volunteers developed vomiting 1 to 3 days after oral administration of a virulent rotavirus strain.54 This was followed by diarrhea lasting 1 to 4 days and associated with anorexia, crampy abdominal pain, and low-grade fever. Viral shedding in stool was detected for 6 to 10 or more days. Two thirds of the adult volunteers developed serologic evidence of infection without disease. Similarly, most naturally occurring rotavirus infections of adults are asymptomatic, manifested only by a rise in antibody titer.55 However, rotavirus infection of adults can also cause severe and even fatal disease.56 Observational studies of children in North America hospitalized with rotavirus gastroenteritis reveal a similar, although more severe, pattern of disease.52,53,57 Rotavirus gastroenteritis in children generally begins with vomiting and fever, which lasts 2 to 3 days, and progresses to profuse watery diarrhea, which continues for 4 to 5 days. Vomiting is more common and prolonged with rotavirus gastroenteritis than with pediatric gastroenteritis caused by most other agents.57 Laboratory findings in children hospitalized with rotavirus gastroenteritis reflect isotonic dehydration and include high urine specific gravity and metabolic acidosis.52,57 Rotavirus gastroenteritis is not generally associated with leukocytosis but is sometimes accompanied by a mild elevation in aminotransferase and uric acid levels.52,57 Liquid stools from children with rotavirus diarrhea usually do not contain blood or fecal leukocytes,52,57 although the presence of fecal leukocytes does not exclude rotavirus as a cause.53 Typically, virus is detected in stools of children by antigenic assays for 4 to 10 days after the onset of symptoms.52 When a sensitive reverse-transcriptase polymerase chain reaction (RT-PCR) assay is used, shedding of viral RNA can be detected for up to 57 days from immunocompetent children, although this does not necessarily indicate shedding of infectious particles.58 Dehydration and severe electrolyte abnormalities leading to cardiovascular failure are the most common proximate causes of death from rotavirus gastroenteritis.59 Seizures and aspiration of vomitus may also lead to death.59 The spectrum of illness after rotavirus infection of infants and children also includes mild gastroenteritis and asymptomatic infection. In some neonatal nurseries, difficult-to-eradicate endemic rotavirus strains asymptomatically infect neonates year round.60,61 A common VP4 type (P[6]) and genetic stability over time suggest that these nursery strains may be less virulent than most circulating strains,60 although maternally transmitted immunity or maturational resistance may also protect the neonates from disease. Because outbreaks of rotavirus diarrhea in neonatal nurseries also occur, any component of maturational resistance cannot be absolute.62 Rotavirus also has extraintestinal manifestations. Viremia, which is common in symptomatic and asymptomatic rotavirus infection, has unknown clinical significance in immunocompetent children.63 In immunocompromised children, rotavirus infection has been associated with chronic diarrhea and extraintestinal infection. Prolonged shedding of live attenuated vaccine strains associated with diarrhea has been observed in infants subsequently diagnosed with severe combined immunodeficiency (SCID), leading to SCID being listed as a contraindication to rotavirus immunization.64 Other conditions associated with chronic rotavirus infection include X-linked agammaglobulinemia, cartilage hair hypoplasia, acquired immunodeficiency syndrome (AIDS), and DiGeorge syndrome.65–68 Rotavirus shed from chronically infected children often has altered genome segments.66,68 Immunohistochemical staining for rotavirus structural and nonstructural proteins in autopsy specimens has demonstrated rotavirus replication in the hepatocytes and renal tubular cells of immunodeficient children with chronic rotavirus infection at the time of death.65 Relatively severe rotavirus gastroenteritis has been noted in adult inpatients with immunosuppression related to bone marrow or renal transplantation.69,70 Rotavirus is not more common or severe in children with HIV disease, and rotavirus immunization is not contraindicated for infants infected with or exposed to HIV.71,72 Rotavirus has been detected in association with a number of syndromes other than gastroenteritis, including respiratory tract infections,73 necrotizing enterocolitis,74 pneumatosis intestinalis,75 myocarditis,76 seizures,77 and meningoencephalitis.77 Because rotavirus infection is universal, these associations may be coincidental rather than causative, and an etiologic association has not been established. Although intestinal intussusception has been temporally associated with immunization with rotavirus vaccines (see “Immunization”), intestinal intussusception does not appear to be associated with natural rotavirus infection.78 Links between rotavirus infection and the subsequent development of autoimmune disease have been proposed. Although some strains of rotavirus can cause biliary atresia in mouse models, and group C rotavirus has been detected in liver tissue from biliary atresia patients, rotavirus has not been convincingly demonstrated to cause this condition in humans.79 Serologic evidence of more frequent rotavirus infection has been associated with the development of celiac disease in predisposed children.80 The introduction of effective rotavirus immunization will clarify whether symptomatic infection with group A rotaviruses is a trigger for such distinctive clinical syndromes. The pathogenesis of rotavirus diarrhea is complex and incompletely understood, with potential roles for a viral enterotoxin, malabsorption related to mucosal damage and depression of disaccharidases, and secretion mediated by the enteric nervous system. Postmortem examination of the gastrointestinal tract of gnotobiotic pigs with diarrhea after experimental infection with a virulent human rotavirus strain demonstrated that virus replicates primarily in the villous epithelium of the small intestine.81 This pattern is consistent with the patchy villous epithelial distribution of rotavirus antigen noted after immunofluorescent staining of duodenal biopsy specimens from children with severe gastroenteritis.82 Light microscopic examination of such duodenal biopsy specimens reveals shortened and blunted villi with a cuboidal epithelium, crypt hypertrophy, and mononuclear cell infiltration of the lamina propria.3,83 The severity of diarrhea in children with rotavirus gastroenteritis correlates with the degree of mucosal damage, which suggests that malabsorption related to loss of absorptive cells may contribute to rotavirus diarrhea late in infection.83 However, in experimentally infected gnotobiotic pigs, diarrhea precedes villous atrophy.81 Similarly, small intestinal biopsy specimens from children with relatively mild rotavirus gastroenteritis do not consistently display histologic changes, probably reflecting patchy epithelial injury.84 These observations indicate that potentially absorptive villous epithelial cells remain despite net fluid losses and that factors other than destruction of the intestinal epithelium are also important in the pathogenesis of rotavirus gastroenteritis. In several animal models, rotavirus infection induces a net secretion of fluid, sodium, and chloride from intestinal segments.85 Glucose cotransport of electrolytes is inhibited,86 although the degree of inhibition in humans does not prevent the use of oral rehydration solutions. Furthermore, decreased disaccharidase activity makes less glucose available for cotransport.3,86 Changes in the molecular weight distribution of absorbed polyethylene glycol in children with rotavirus gastroenteritis indicate an increase in paracellular permeability.87 Experiments in cell culture suggest that disruption of tight junctions by VP8* or NSP4 may mediate this increased permeability.88,89 The rotavirus nonstructural glycoprotein NSP4 is an enterotoxin that causes diarrhea in some animal models.90 The fragment of NSP4 associated with this activity forms oligomers through a tetrameric or pentameric coiled-coil and raises cytoplasmic calcium levels, leading to a secretory state by activating a calcium-dependent chloride channel of the TMEM16 family.91–94 Several mechanisms have been proposed for NSP4’s effect on cytoplasmic calcium levels. Most NSP4 is resident in the ER membrane, and residues 47-90 have a viroporin-like activity that could disrupt the ER membrane, releasing calcium stores in infected cells.95 Alternatively, an NSP4 peptide corresponding to residues 112-175 and intact NSP4 in lipoprotein complexes are released from rotavirus-infected cells and could affect uninfected cells.96,97 Extracellularly applied NSP4 fragments stimulate phospholipase C to produce inositol-1,4,5-triphosphate, which leads to calcium influx and release of calcium from intracellular stores.98 Other mechanisms proposed for NSP4 enterotoxin activity include inhibition of sodium absorption by the epithelial sodium channel ENaC and by the glucose-coupled sodium cotransporter SGLTI and interference with lactase function.94,99,100 Although orally administered antibodies against NSP4 protect mice from rotavirus diarrhea, actively acquired anti-NSP4 antibody fails to protect gnotobiotic piglets from challenge, and the role of an extracellular enterotoxin in human rotavirus gastroenteritis is not yet established.90,101 The enteric nervous system has a role in the pathogenesis of rotavirus gastroenteritis. Treatment of rotavirus-infected mice with lidocaine (a sodium channel–blocking anesthetic), granisetron (a serotonin receptor antagonist), or a vasoactive intestinal peptide receptor antagonist attenuates diarrhea.102,103 The ability of racecadotril, an enkephalinase inhibitor, to attenuate rotavirus diarrhea in children confirms that enteric nervous system–mediated secretion contributes to the pathogenesis of diarrhea in humans and is a target for therapeutic intervention (see “Therapy”).104 Secretion of nitric oxide, a mediator of intestinal motility, from NSP4-exposed human epithelial intestinal cells (HT-29) in culture and secretion of serotonin from an NSP4-exposed human midgut carcinoid enterochromaffin cell line (GOT1) suggest a potential role for the enteric nervous system in rotavirus-mediated intestinal hypermotility and vomiting.105,106 The diversity of rotaviruses, their ability to exchange genome segments encoding antigenic determinants, and changing diagnostic technology have led to evolving serology and classification systems. Rotavirus strains have been classified serologically into serogroups, subgroups, G serotypes, and P serotypes and genetically into electropherotypes, genogroups, G genotypes, and P genotypes. To organize the increasing volume of whole-genome rotavirus sequence data, in 2008 the Rotavirus Classification Working Group (RCWG) developed a nucleotide sequence–based complete genome classification system for group A rotaviruses.107 The broadest serologic designation is the serogroup. The seven serogroups, A through G, are defined by the cross-recognition of particles by serum antibody obtained from parenterally hyperimmunized animals.108 Serogroup determinants are predominantly located on VP2 and VP6 (the innermost and middle layer proteins). Genetic exchange has not been observed between members of different serogroups. Serogroups A, B, and C cause disease in humans and other animals; groups D through G have been described only in nonhuman animals. Serogroup A is the most important clinically, as group A viruses cause the endemic gastroenteritis of children; groups B and C have been associated with epidemics of gastroenteritis affecting all ages. Group B includes the strain ADRV (adult diarrhea rotavirus), which has been associated with large outbreaks of severe diarrhea in adults in China.109 Group C viruses have been associated with less severe gastroenteritis in both children and adults.110,111 Serogroup A is subdivided into subgroups I, II, I+II, and non-I non-II on the basis of monoclonal antibody recognition of antigenic determinants on VP6.112 Within group A, rotavirus serotypes are defined on the basis of reciprocal cross-neutralization by antibody. Initially, rotavirus serotypes were designated by a unitary system, which primarily reflected neutralizing antibodies directed against VP7. This system did not fully explain cross-neutralization patterns because VP4 also contains neutralization determinants.113,114 Group A rotaviruses are now classified into serotypes with a binomial nomenclature, in which neutralization by antibodies against VP7 defines “G” serotype (for glycoprotein antigen) and neutralization by antibodies against VP4 defines “P” serotype (for protease-sensitive antigen). Further complicating the serology, VP7 and VP4 are each targets both of homotypic antibodies that neutralize only within serotype and of heterotypic antibodies that neutralize several serotypes.114,115 Serotype-specific epitopes of VP4 are predominantly found on the variable VP8* hemagglutination domain, at the tips of the spikes, and heterotypic epitopes are predominantly found on the more conserved β-barrel domain, in the body and stalk of the spikes (see Fig. 152-2B, left).10,12,29 Immunodominant serotype-specific and heterotypic neutralizing epitopes of VP7 straddle the calcium-containing interface between the subunits of each VP7 trimer, and additional neutralizing epitopes are found at an interdomain boundary within each VP7 subunit.26 Early attempts at genetic classification grouped rotaviruses according to RNA genome electropherotype, which distinguished group A strains as “long,” “short,” and “supershort” on the basis of the electrophoretic mobility of genome segments 10 and 11. Whole rotavirus genome analysis indicates that there are two main genogroups of human rotaviruses, each of which contains viruses with overall genome similarity.116 The Wa-like human rotavirus genogroup appears to share origins with porcine rotaviruses, and the DS-1-like genogroup appears to share origins with bovine rotaviruses.116 In the RCWG genetic classification, a separate genotype is assigned to each of the 11 genome segments. Gx, P[x], Ix, Rx, Cx, Mx, Ax, Nx, Tx, Ex, and Hx designate the genes that encode VP7, VP4, VP6, VP1, VP2, VP3, NSP1, NSP2, NSP3, NSP4, and NSP5/6, respectively.107 The RCWG has proposed a strain-naming convention in which strains are named “RV serogroup/species of origin/country of identification/common name/year of identification/G- and P-type.”117 Because the sequence of the genome segment encoding VP7 accurately predicts G serotype, genotyping provides a practical surrogate for G serotype determination. As of April 2014, the web-based database maintained by the RCWG (http://rotac.regatools.be/) listed 27 G types. Of these, five (G1-4 and G9) comprise 88% of human rotavirus isolates from 1996 to 2007.118 To date, at least 14 P serotypes (with serotypes 1, 2, and 5 each divided into subtypes with letter designations, such as P1A) have been identified.107,118 Immunologic reagents for determining P serotype are limited, and P serotyping is complicated by cross-reactivity between P serotypes. Therefore, P genotyping is more commonly used for classification. As of April 2014, 37 P genotypes (P[1] to P[37]) were listed in the RCWG database. Unlike G serotypes, which have a one-to-one correspondence with G genotypes, some P serotypes include more than one P genotype. In strain descriptions, the genotype is enclosed in brackets after the serotype designation. Thus, G2P1B[4] refers to a virus of G serotype 2, P serotype 1B, and P genotype 4. Two P genotypes, P[4] and P[8], cause most human rotavirus disease. Indeed, despite the diversity of rotavirus genotypic combinations, just five pairings, G1P[8], G2P[4], G3P[8], G4P[8], and G9P[8], contain approximately 75% of human rotavirus isolates from 1996 to 2007.117 Substantial geographic and temporal variation exists in strain distribution. For example, G8 rotavirus strains are prevalent in high rotavirus mortality countries in Africa and therefore have more clinical significance than their global prevalence would suggest.117 Two novel genotypic combinations, G9P[8] and G9P[6], have spread globally since the mid-1990s.117 Reassortment between human and animal rotavirus strains, antigenic drift, and the occasional introduction of animal rotaviruses into the pool of viruses circulating among humans provide a continuous introduction of genetic diversity, necessitating ongoing surveillance to determine whether rotavirus vaccines will require strain changes in the future for continued efficacy.116 Evidence is emerging of an increased prevalence of G2P[4] viruses in regions with high coverage by a G1P[8] monovalent rotavirus vaccine, suggesting that immunization may start to alter the genotype distribution of circulating strains.119 Rotavirus infection is universal, and almost all children acquire serum antibody against the virus in the first 2 or 3 years of life.120 Severe gastroenteritis caused by rotavirus most commonly affects infants and children between 6 months and 2 years of age,121 although in lower socioeconomic populations the peak of illness may be somewhat earlier.121–123 The age at greatest susceptibility to severe disease may be bracketed by the waning of maternally transferred passive immunity and the maturation of the gastrointestinal tract at about 6 months and by the acquisition of active immunity related to natural infection later in childhood. Rotavirus is the most common cause of severe dehydrating diarrhea leading to the hospitalization of infants and children in both the developed and developing world. Analyses of published studies indicate that, as of 2008, rotavirus caused 453,000 childhood deaths each year worldwide, corresponding to 37% of diarrhea-related deaths and 5% of all deaths for infants and children younger than the age of 5 years.1 The incidence of rotavirus gastroenteritis is similar in developed and developing countries, but mortality is concentrated in poorer nations (Fig. 152-3).1 The causes of higher mortality from rotavirus in the developing world may include the greater prevalence of malnutrition, less access to treatment, larger inoculum size, and synergy with other intestinal flora or pathogens.122 Global mortality rates from rotavirus infection appear to have fallen by approximately 50% from 1985 to 2008, consistent with an overall reduction in mortality from diarrhea, although methodologic differences between studies make the exact amount of decline uncertain.1,124 Improved access to treatment, particularly increased use of oral rehydration solutions, probably plays a role in the lower mortality rates. Better nutrition and a lower incidence of comorbidities may also contribute.
Rotaviruses
History and Overview
Viral Structure and Replication
Clinical Manifestations
Pathogenesis
Serologic Classification
Epidemiology
Rotaviruses
152