Role of the Endothelium in Hemostasis
Michael J. Krisinger
Edward M. Conway
Hemostasis and coagulation, which refer to the arrest of bleeding and the formation of a fibrin clot, respectively, are generally used interchangeably to describe the processes by which blood is maintained in a fluid state in the circulatory system. Normal function of the endothelium, the single cell layer lying at the interface of circulating blood and the underlying tissue, is required for optimal organ function and for maintaining and regulating blood flow to meet the body’s dynamically changing demands over a range of pathophysiologic conditions. Rather than playing a passive role in the transport of blood, the endothelium has, in the past half century, become recognized as a central and active player in homeostasis. In response to damaging stimuli, such as trauma or infections, the vascular endothelium, in conjunction with other components of the hemostatic system, serves to rapidly and effectively limit tissue destruction to a local site and to promote healing.
The hemostatic system is continuously in an “on” position, dynamically exhibiting antithrombotic and prothrombotic properties that are delicately balanced to meet local and systemic needs (FIGURE 43.1). Under normal physiologic conditions, natural antithrombotic forces override the prothrombotic ones, limiting platelet adhesion and activation and excess fibrin clot formation, while vasomotor tone is optimized, overall maintaining critical blood flow to ensure organ function. Not unexpectedly, the switches that control hemostatic system activity are tightly regulated. Genetic, environmental, and epigenetic factors that disrupt the exquisite and dynamic balance between prothrombotic and antithrombotic forces are clinically manifest by either bleeding or thrombosis. The major prothrombotic forces are mediated by platelets, monocytes, the clotting cascade, and the vascular endothelium, while the predominant antithrombotic mechanisms center around the endothelium. In this chapter, we explore the major properties of the endothelium in regulating the hemostatic system.
TISSUE FACTOR AND INITIATION OF COAGULATION ON THE ENDOTHELIAL CELL SURFACE
The coagulation system is characterized by the sequential activation of a series of serine proteases, culminating in the generation of thrombin, with subsequent conversion of fibrinogen into an insoluble fibrin clot. The molecular events surrounding initiation of coagulation have been extensively examined and revised since the original description in 1964 of the cascade/waterfall hypothesis of hemostatic system activation.1 Following vascular injury, tissue factor (TF) is exposed to the circulation and binds to coagulation factor VIIa.2 When not associated with TF, the factor VIIa that constitutively circulates at low levels has poor enzymatic properties. When complexed with TF, factor VIIa acquires potent catalytic properties and triggers coagulation by converting the zymogens factor IX and factor X to enzymes factor IXa and factor Xa, respectively. The initial factor Xa formed when bound to membrane is sufficient to generate trace (pM) amounts of thrombin from prothrombin. This initial thrombin serves as the primary activator of the zymogen factor XI, the procofactors V and VIII, and importantly platelets, which together provide important amplification feedback. Surface exposure of anionic phospholipids (e.g., phosphatidylserine) is essential in promoting assembly and efficient catalytic activity of the two subsequent membrane-bound enzyme complexes, namely “intrinsic factor Xase” and “prothrombinase.” The factor IXa generated from the TF-factor VIIa combines with its cofactor factor VIIIa on the activated platelet membrane to form the “intrinsic factor Xase,” which becomes the major activator of factor X. Similarly, the factor Xa formed combines with factor Va on the activated platelet membrane to form “prothrombinase,” which converts prothrombin to thrombin.
In what is viewed as the central event in coagulation, the serine protease thrombin finally cleaves fibrinogen to generate fibrin, which is cross-linked by factor XIIIa. The scheme supports the notion that the TF-factor VIIa pathway of blood clotting is the major physiologic mechanism for triggering coagulation and that exposure of TF to the circulation is the critical event that induces the vasculature to acquire a procoagulant phenotype.3
TF is a 263-amino acid integral membrane protein, structurally comprised of three domains.4 The extracellular domain constitutes over 80% of the amino acid sequence of the molecule and provides binding sites for factor VIIa, such that the protease activity of TF-complexed factor VIIa toward its substrates, factors X and IX is dramatically increased. The transmembrane domain of TF is important for optimizing its cofactor function with factor VIIa.5 The cytoplasmic tail mediates intracellular signals in processes that impact on a range of cellular activities, including cell proliferation, angiogenesis, and inflammation.6,7,8
Expression of TF varies in different organs, with particularly high levels in the heart, lung, brain, testis, uterus, and placenta, and lower amounts in the joints and skeletal muscle.9 It is found throughout the vasculature, synthesized by adventitial fibroblasts, smooth muscle cells, pericytes, and several hematopoietic and nonhematopoietic cells.10,11 When stimulated by proinflammatory cytokines, monocytes and macrophages prominently express TF.10,12 Tumor cells also synthesize TF, which impacts on metastasis formation.13 In some pathologic conditions, small amounts of TF that are in or on platelets and/or a component of plateletderived microparticles may also contribute to the overall pool of TF, although the relevance of these sources is not yet clear.14,15
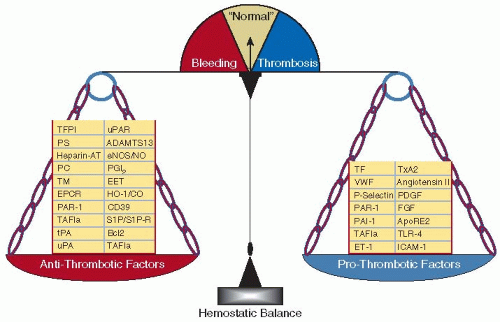 FIGURE 43.1 Balance between endothelial prothrombotic and antithrombotic factors. Relative imbalances in expression of prothrombotic and antithrombotic factors will determine the response to pathophysiologic stresses that may be exhibited by bleeding or thrombosis. |
In spite of some conflicting data, most studies concur with the earliest reports that TF is not normally expressed by endothelial cells in vivo.16,17 Indeed, in the absence of injury, functionally active TF is excluded from the intravascular space. However, during disease, vascular endothelial cells may express TF. For example, TF may be prominently expressed by endothelial cells of the pulmonary veins of hypoxic mice with sickle cell anemia,18 of tumor vessels,19 and overlying atheromatous lesions.20 Some investigators have suggested that TF is normally expressed by circulating monocytes but in a functionally inactive “encrypted” form, able to undergo “decryption” during inflammation.21 Thus, given its prominent perivascular distribution, TF has been represented as a hemostatic “envelope” for the vasculature, readily available to initiate coagulation upon exposure to blood when vascular integrity is compromised by chemical or mechanical injury.16 Indeed, recent studies suggest that preformed TF-factor VIIa complexes may reside around blood vessels in the absence of injury, allowing even more rapid coagulation activation upon injury.22
Numerous extracellular stimuli that are implicated in disorders associated with thrombosis may trigger coagulation in vivo by inducing TF expression by most cells exposed to the circulation, including for example endothelial cells, monocytes, platelets, and subendothelial cells (after vessel injury). Among these agonists are proinflammatory mediators, including lipopolysaccharide, tumor necrosis factor (TNF)-α, interleukin (IL)-1β, thrombin, vascular endothelial growth factor (VEGF), infectious agents (e.g., herpes simplex virus, rickettsia, and measles), oxygen free radicals, and low shear stress. This list is not comprehensive, but underlines the fact that the vascular endothelium is constantly exposed to agents that are likely to induce varying degrees of damage with exposure of TF, with constant generation of small amounts of thrombin. Indeed, it has been well established that in normal individuals, there is a persistent low level of activation of coagulation.23,24 For the TF-factor VIIa triggering reaction to proceed to full-fledged coagulation, the amount of TF exposed and thus the amount of thrombin generated must exceed a threshold to overcome the activity of endogenous natural anticoagulant pathways that otherwise serve to limit thrombin bursts and prevent physiologically excessive thrombotic responses. There are in fact, four dominant natural vasculoprotective mechanisms that regulate thrombin generation and fibrin clot formation. These are tissue factor pathway inhibitor (TFPI), the heparin-antithrombin (AT) pathway, the protein C (PC) anticoagulant pathway, and the fibrinolytic system.
TISSUE FACTOR PATHWAY INHIBITOR
TFPI-1, commonly referred to as TFPI, is the principle inhibitor of the TF pathway, binding to and neutralizing TF-factor VIIa in a factor Xa-dependent manner (reviewed in25,26). TFPI is primarily associated (-85%) with microvascular endothelial cells where it is constitutively expressed. Lesser amounts are also found on the surface of monocytes, smooth muscle cells, platelets, mesangial cells, fibroblasts, cardiomyocytes, and plasma.27,28
TFPI is a 276-amino acid, 43 kDa, multivalent Kunitz-type serine protease inhibitor29 that comprises an acidic N-terminal region, three Kunitz-type domains, and a basic C-terminus. Interaction of TFPI with the endothelial cell surface is achieved via two mechanisms. The C-terminal stretch of basic residues of TFPI binds to cell surface glycosaminoglycans.27 This is therefore heparin releasable and accounts for a two- to fourfold increase in plasma levels after heparin infusion.30 Most of the cell surface-associated TFPI, which cannot be released by heparin, is bound through a glycosylphosphatidylinositol (GPI) anchor.31 However, TFPI does not contain a consensus sequence for attachment of a GPI anchor, and thus it is believed that there exists a hitherto unidentified coreceptor that binds to the third Kunitz domain of TFPI.
Alternative pre-mRNA splicing yields at least three isoforms of TFPI in the mouse (α,β, ρ) and human (αβο). Only the fulllength form (TFPIα or TFPI) contains the third Kunitz domain and the basic C-terminal region. The other isoforms contain
two Kunitz domains and the C-terminal region. In humans, TFPI is the only one that has been identified at the protein level in tissues.25 During embryonic development in mice, TFPI and TFPIβ are both widely expressed and at about equal levels. However, adult mouse tissues produce mostly TFPIβ. In contrast to TFPI, TFPIβ has a C-terminal GPI anchor attachment sequence, allowing it to directly associate with the cell surface.31 The role of TFPIρis unknown. Although it is believed that inhibition of TF-factor VIIa and factor Xa require the third Kunitz and the basic C-terminus of TFPI, several in vitro studies suggest that TFPIβ may have anticoagulant activity on the cell surface.32 In vivo studies are required to fully assess the relative anticoagulant activity of TFPIβ. Functional data for TFPIο are lacking. It contains two Kunitz domains and a 12-amino acid C-terminus, the latter having no similarity to the other isoforms.
two Kunitz domains and the C-terminal region. In humans, TFPI is the only one that has been identified at the protein level in tissues.25 During embryonic development in mice, TFPI and TFPIβ are both widely expressed and at about equal levels. However, adult mouse tissues produce mostly TFPIβ. In contrast to TFPI, TFPIβ has a C-terminal GPI anchor attachment sequence, allowing it to directly associate with the cell surface.31 The role of TFPIρis unknown. Although it is believed that inhibition of TF-factor VIIa and factor Xa require the third Kunitz and the basic C-terminus of TFPI, several in vitro studies suggest that TFPIβ may have anticoagulant activity on the cell surface.32 In vivo studies are required to fully assess the relative anticoagulant activity of TFPIβ. Functional data for TFPIο are lacking. It contains two Kunitz domains and a 12-amino acid C-terminus, the latter having no similarity to the other isoforms.
To effectively regulate TF-factor VIIa-induced initiation of coagulation, TFPI must be appropriately localized at the cell surface where vascular injury exposes TF to the circulation. At that site, TFPI is well positioned to prevent small amounts of TF-factor VIIa, formed in response to a minor injury, from unnecessarily generating a large thrombin burst. Indeed, TFPI provides the first natural barrier threshold, without which coagulation would otherwise proceed and amplify in an unregulated fashion (FIGURE 43.2). TFPI utilizes the two N-terminal (first and second) Kunitz-type serine protease domains to inhibit the active sites of both factor Xa and TF-factor VIIa. As TF-factor VIIa activates factor X, the second Kunitz domain of TFPI binds to the active site of the newly generated factor Xa that is still associated with the TF-factor VIIa complex, rendering the factor Xa inactive. The flanking Kunitz domains and the basic C-terminal region of TFPI enhance their interaction with factor Xa. Protein S (PS), a vitamin K-dependent protein and cofactor in the PC-thrombomodulin (TM) anticoagulant system (see below), also acts as a cofactor for TFPI by interacting with its third Kunitz domain and its C-terminus, accelerating inhibition of factor Xa.33,34 Binding of factor Xa causes TFPI to undergo a conformational change, thereby facilitating subsequent interaction of the TFPI-factor Xa complex via the first Kunitz domain of TFPI, in a Ca2+-dependent manner, with the active site of factor VIIa when bound to TF.35 The resultant quaternary complex TF/factor VIIa/factor Xa/TFPI lacks both TF-initiated blood coagulation activity as well as protease-activated receptor (PAR)-1/2-mediated cellular signaling activity involved in inflammation. Although the interactions of TFPI with factor Xa and TF-factor VIIa are usually depicted as a two-step process, these effectively occur simultaneously. Indeed, once factor Xa is released from TF-factor VIIa and complexed with factor Va, it cannot any longer be inhibited by TFPI.36 Thus, the window during which TFPI must function is critically narrow.
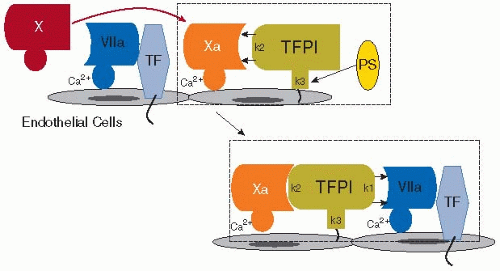 FIGURE 43.2 TFPI-mediated neutralization of factors VIIa and Xa. Localized to the site of injury on the endothelial cell surface, tissue factor pathway inhibitor (TFPI) binds first to factor Xa via its second Kunitz domain (k2), enhanced by protein S (PS) which interacts with the C-terminus and the third Kunitz domain (k3) of TFPI. TFPI then undergoes a conformational change, facilitating Ca2+-dependent binding via the first Kunitz domain (k1) and neutralization of factor VIIa which is complexed with tissue factor (TF). |
As noted above, TFPI has several cellular sources. While endothelial TFPI predominates, it is synthesized by megakaryocytes, and stored in α-granules of platelets, whereupon it may be translocated to the platelet surface upon activation.37 TFPI is also found in the plasma, the origin of which is primarily endothelial. It circulates in a complex with low-density lipoproteins (LDLs), and comprises various degraded forms, probably generated by proteolysis by a range of enzymes, including thrombin, plasmin, and matrix metalloproteinases.26 The gamma-carboxyglutamic acid (Gla) domain of PS and the C-terminal region of TFPI effectively bind negatively charged phospholipid surfaces and thus localize this negative regulatory system to sites where TF may be exposed for initiation of coagulation, including injured or denuded endothelial cells and activated platelets.33,34 Nonetheless, delineation of the role of different pools of TFPI and their physiologic site(s) of action require further study.
Not surprisingly, inactivation of the TFPI gene in mice results in embryonic lethality38 due to a consumptive coagulopathy. Heterozygous TFPI deficiency in combination with factor VLeiden in mice increases thrombotic susceptibility.39 The finding that TFPI administration had efficacy in animal models of sepsis prompted phase III clinical trials. Unfortunately, although TFPI dampened activation of coagulation in patients with sepsis, there was no survival benefit.40 Indeed, the link in humans between TFPI deficiency and thrombotic disease has not been clearly established. There are suggestions that inadequate release of TFPI into the circulation might increase the risk of thrombosis, and only isolated TFPI polymorphisms have been weakly associated with thrombosis.41 This lack of a clear association is intriguing, as it implies that there may be additional natural anticoagulant mechanisms that compensate for deficiencies in TFPI. It may also mean that moderate deficiencies of TFPI may be incompatible with life.
Similar to TFPI (TFPI-1) in structural architecture, TFPI-2 also has three Kunitz-type domains, but does not inhibit factor Xa and has minimal inhibitory activity for TF-factor VIIa.42,43 Rather, TFPI-2 expressed by endothelial cells, inhibits trypsin, plasmin, kallikrein, factor XIa, and some matrix metalloproteinases, interacts with complement-binding proteins, and
suppresses growth factor-induced proliferation of endothelial cells.44 Although TFPI-2 may play a role in the coagulation cascade and as a vasculoprotective molecule, it is more likely important in inflammation, atherosclerosis, and tumor cell invasion.45
suppresses growth factor-induced proliferation of endothelial cells.44 Although TFPI-2 may play a role in the coagulation cascade and as a vasculoprotective molecule, it is more likely important in inflammation, atherosclerosis, and tumor cell invasion.45
ANTITHROMBIN AND ENDOTHELIAL HEPARIN SULFATE
AT is a circulating serine protease inhibitor (serpin) that neutralizes thrombin, factor Xa and factor IXa,46 and to a lesser extent the procoagulant factors XIa, XIIa, and TF-factor VIIa. It also exhibits some activity against plasmin, kallikrein, urokinasetype plasminogen activator (uPA), and tissue-type plasminogen activator (tPA). AT is synthesized primarily in the liver and has a molecular weight of approximately 58 kDa. In the circulation, AT exists in a “repressed” conformation that has minimal protease inhibitory activity. In the presence of heparin or heparin sulfate, the neutralizing capacity of AT is accelerated over 500-fold for factor Xa, more than 2,000-fold for thrombin, and approximately one millionfold for factor IXa. Indeed, this is the rationale for using heparin as an anticoagulant drug. Endogenous heparin, however, does not freely circulate in the blood. Rather, endothelial cells express heparin sulfate-containing proteoglycans, including, for example, syndecan-1, -2, -4, glypican-1, betaglycan, CD44, and fibroblast growth factor (FGF) receptor-2, allowing direct interactions with AT,47,48 as well as with TFPI, thrombin, heparin cofactor II, and tPA. The pattern of sulfation on the repeating disaccharide backbone helps define the binding specificity of each heparin sulfate structure. Although somewhat controversial,49,50 it is generally believed that the anticoagulant properties of AT are localized to the surface of the endothelium, thereby contributing to the nonthrombogenic properties of the vessel wall. Vascular-bed-specific heterogeneity in the expression of the different heparin sulfate-containing proteoglycans helps to explain the diversity in the anticoagulant properties within different vascular beds.51,52 In the setting of injury, luminal and subluminal sources of heparin may also activate AT, thereby restricting clot extension.
Heparin-induced activation of AT to a form that rapidly inhibits procoagulant serine proteases occurs via at least two mechanisms (FIGURE 43.3). Through ionic and nonionic interactions, a sequence-specific, negatively charged pentasaccharide within heparin binds to AT with high affinity, thereby inducing allosteric changes in AT that allow it to specifically react with and inhibit factors Xa and IXa. This pentasaccharide is insufficient but necessary to promote inhibition of thrombin by AT. The pentasaccharide must be part of a longer negatively charged polysaccharide of heparin (minimallya 16-mer), which provides a binding site for thrombin. With AT simultaneously bound to the pentasaccharide and a more remote polysaccharide chain, the thrombin molecule thus can be oriented via this heparin bridging mechanism for neutralization by AT. This bridging effect also enhances AT reactivity with factors Xa and IXa for more effective neutralization.53,54,55 Protease inhibition rates by AT due to heparin sulfate bridging and allosteric changes in AT are estimated to be augmented approximately 2,000-fold and 300-fold, respectively.56,57 Interestingly, heparin-activated AT is a poor neutralizer of the natural anticoagulant serine protease, activated PC (APC), highlighting the specificity of AT for procoagulant proteases.58
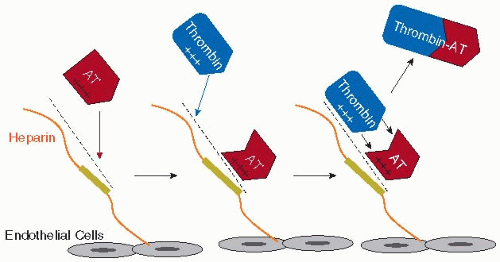 FIGURE 43.3 Heparin-AT neutralization of thrombin. Antithrombin (AT) binds, via nonionic and ionic interactions, to a specific negatively charged pentasaccharide (green) within heparin or heparan sulfate molecules on the endothelial cell surface and undergoes a conformational change, allowing it to efficiently react with and inactivate factors Xa and IXa (not shown). For AT neutralization of thrombin, a longer polysaccharide chain provides an additional binding site for thrombin (exosite II), bridging the interaction between thrombin and AT. Once formed, the thrombin-AT complex dissociates from the polysaccharide chain and is cleared from the circulation. |
AT also plays a role in protecting organisms against inflammatory and infectious disorders, in part via effects on the vascular endothelium. At least in animal models, administration of AT protects against septic shock,59 although this has not uniformly been observed in human studies.60 In vitro, AT interferes with nuclear factor kappa B (NF-kB) nuclear translocation, thereby suppressing release of cytokines by endothelial cells and the expression of leukocyte adhesion molecules. AT also enhances prostacyclin (PGI2) release from the vasculature61 that was protective in a rat model of abdominal sepsis.62
The physiologic importance of the heparin-AT mechanism as a natural inhibitor of coagulation is unquestioned. Inherited heterozygous AT deficiency has a reported prevalence that ranges from 1:500 to 1:5,000 and is associated with a significantly increased risk of venous thromboembolic disease.63,64 Indeed, by the age of 50 years, approximately 50% of patients with AT deficiency will have had at least one thrombotic episode, half of which will be unprovoked.65 Congenital or inherited deficiencies of heparin have not been reported, suggesting that its absence is not compatible with survival.
THE PROTEIN C SYSTEM
The PC system represents a major physiologically relevant natural anticoagulant mechanism, the function of which depends on the integrity of endothelial cell integral membrane glycoproteins TM and endothelial protein C receptor (EPCR). This system also provides a critical link between coagulation and
inflammation,66 highlighting how these biologic processes are spatiotemporally integrated.
inflammation,66 highlighting how these biologic processes are spatiotemporally integrated.
PC is a vitamin K-dependent protein, synthesized in the liver that circulates as a two-chain zymogen.67 APC is a serine protease that is generated by thrombin-mediated cleavage of PC, an event that requires TM, and is indeed amplified over 1,000-fold by its presence68 (FIGURE 43.4). As the name implies, TM has the remarkable ability to modulate thrombin by allosteric alteration of its active site, converting it from a procoagulant enzyme to an anticoagulant enzyme. This switch in substrate specificity prevents thrombin-TM from cleaving fibrinogen, activating factor V and factor VIII, and triggering platelet activation. PC activation by thrombin-TM is amplified a further 20-fold when PC is bound to EPCR.69 Platelet factor 4 (PF4) may also accelerate PC activation by inducing allosteric changes in PC that increase its affinity for thrombin-TM.70 Facilitated by its cofactor PS, APC has potent anticoagulant activities by proteolytically cleaving and inactivating factors VIIIa and Va, which thereby suppresses further production of factor Xa and thrombin71,72 (FIGURE 43.5). The physiologic importance of this system is underlined by the fact that patients with deficiencies of PC or PS, or who have a mutation in factor V (factor VLeiden) that renders it resistant to inactivation by APC, all have an increased risk of thromboembolic disease73,74,75 APC also has profound anti-inflammatory properties, mediated by several mechanisms including, for example, suppressing exposure of TF and the release of TNF-α, IL-1β, IL-6, and IL-8, and preventing endothelial expression of leukocyte adhesion molecules.76,77,78 Numerous in vivo studies have confirmed the important antiinflammatory properties of APC.6
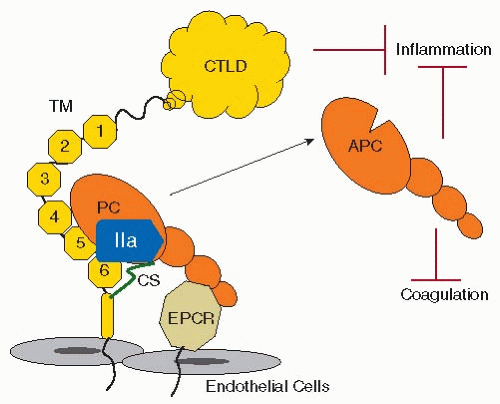 FIGURE 43.4 Activation of PC by thrombin-TM-EPCR. EGF-like 4-6 repeats of thrombomodulin (TM, yellow), the chondroitin sulfate (CS) of TM, and the endothelial protein C receptor (EPCR) provide cofactor function for thrombin (IIa)-mediated activation of protein C (PC). Activated protein C (APC) has anticoagulant and antiinflammatory properties. The C-type lectin-like domain (CTLD) of TM also suppresses inflammation. |
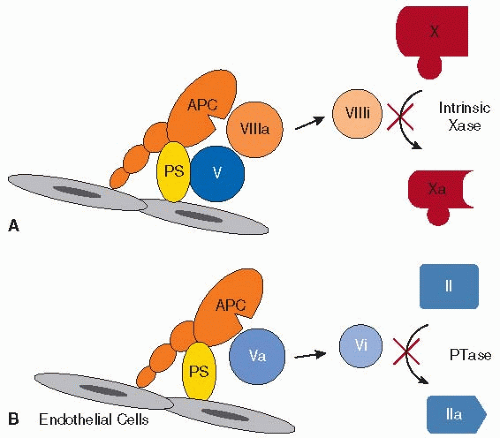 FIGURE 43.5 Anticoagulant properties of APC. When free of EPCR, APC localizes with cofactor protein S (PS) on the surface of endothelial cells or platelets and inactivates factor VIIIa, using factor V as a synergistic cofactor (A) or inactivates factor Va (B). This yields, respectively, the inactive species factor VIIIi and factor Vi. Lack of the active cofactors (factors VIIIa and Va) abrogates conversion of factor X to factor Xa by the intrinsic factor Xase complex (A), and prothrombin (II) to thrombin (IIa) by the prothrombinase (PTase) complex (B), respectively. |
The thrombin-TM complex exhibits additional effects on hemostasis through the activation of the plasma procarboxypeptidase, thrombin-activatable fibrinolysis inhibitor (TAFI). Activation of TAFI by thrombin alone is inefficient but in the presence of TM, is enhanced over 1,000-fold79 (FIGURE 43.6). The carboxypeptidase activity of activated TAFI (TAFIa), in turn, cleaves C-terminal lysine residues from fibrin cleavage products.80 Fibrin cleavage products, produced by limited plasmin proteolysis of fibrin, accelerate the conversion of plasminogen to plasmin by tPA. Thus, removal of these C-terminal lysine residues impedes the degradation of insoluble fibrin, with less incorporation of plasminogen and tPA into the fibrin clot. Accordingly, TM serves an important antifibrinolytic function. TM may also be considered a profibrinolytic factor, since, via APC, it suppresses thrombin generation which
in turn, down-regulates further TAFIa production. Patients with impaired thrombin generation due to factor XI deficiency exhibit a bleeding tendency that is contributed to by excess fibrinolysis that may be attributed to reduced thrombin-TM-mediated activation of TAFI.81 Extensive studies have been performed in mice lacking TAFI. However, the in vivo significance of TAFIa as a regulator of fibrinolysis remains controversial (reviewed in82), although elevated circulating levels of TAFIa in humans may confer a mild increased risk of thrombosis.83 A role for TAFIa as a natural anti-inflammatory molecule has more recently become of greater interest with recognition of its ability to inactivate the anaphylatoxins C3a and C5a by des-argination.84 Moreover, the proinflammatory mediators, bradykinin and osteopontin,85 are also inactivated by TAFIa.
in turn, down-regulates further TAFIa production. Patients with impaired thrombin generation due to factor XI deficiency exhibit a bleeding tendency that is contributed to by excess fibrinolysis that may be attributed to reduced thrombin-TM-mediated activation of TAFI.81 Extensive studies have been performed in mice lacking TAFI. However, the in vivo significance of TAFIa as a regulator of fibrinolysis remains controversial (reviewed in82), although elevated circulating levels of TAFIa in humans may confer a mild increased risk of thrombosis.83 A role for TAFIa as a natural anti-inflammatory molecule has more recently become of greater interest with recognition of its ability to inactivate the anaphylatoxins C3a and C5a by des-argination.84 Moreover, the proinflammatory mediators, bradykinin and osteopontin,85 are also inactivated by TAFIa.
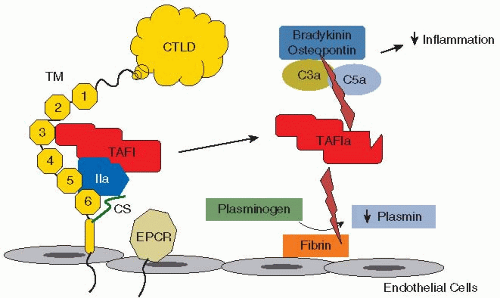 FIGURE 43.6 Activation of TAFI by thrombin-TM. EGF-like 3-6 repeats of TM provide cofactor function for thrombin-mediated generation of TAFIa, which cleaves basic C-terminal amino acid residues from several substrates including complement components C3a and C5a, bradykinin, and osteopontin, thereby reducing proinflammatory forces. TAFIa also cleaves fibrinogen, interfering with efficient generation of plasmin from plasminogen. |
TM, encoded by an intronless gene, is a 557-amino acid, type 1 transmembrane glycoprotein (GP). It is expressed by endothelial cells in all vascular beds. The ectodomain of TM comprises a juxtamembranous serine/threonine-rich domain with potential sites for O-linked glycosylation which support the attachment of a chondroitin sulfate that enhances the activation of PC by thrombin-TM,86 accelerates the neutralization of thrombin by heparin-AT, and facilitates binding of PF4 to PC to increase its activation. The next domain contains six epidermal growth factor (EGF)-like repeats. EGF-like repeats 3, 4, 5, and 6 (EGF3-6) are essential for thrombin-mediated activation of PC and TAFI.87,88,89,90 Via its anion binding exosite I, thrombin binds to EGF5-6. EGF4-6 is required for activation of PC,91 and EGF3-6 is required for TAFI activation.92 An aspartic acid residue (Asp349) within EGF4 is required for Ca2+-mediated binding of TM to the gamma-carboxyglutamic acid (Gla) containing domain of PC, and thus for activation of PC by thrombin. Oxidation of methionine 388, which resides between EGF4 and EGF5, abrogates thrombin’s ability to activate PC, an event that likely occurs during inflammation when neutrophils are activated. At the N-terminus of the molecule, there is a globular C-type lectin-like domain.93 This structure lacks anticoagulant function, but plays a major role in inflammation (FIGURE 43.4). Mice lacking the lectin-like domain of TM are more sensitive to endotoxin challenges and proinflammatory stimuli and exhibit increased tissue accumulation of neutrophils and monocytes, and excess activation of complement,94 in part by preventing the proinflammatory cytokine high-mobility group box 1 from binding to the receptor for advanced glycosylation end products.95
As might be expected, endothelial TM expression is reduced in many prothrombotic conditions. This is achieved via transcriptional and posttranscriptional mechanisms. Thus, for example, shear stress,96 oxidized LDL,97 hyperhomocysteinemia,98 proinflammatory cytokines,99,100 sepsis,101 and aging102 are associated with suppressed TM levels and often enhanced TF exposure, an imbalance that promotes fibrin deposition. Conversely, TM is upregulated by VEGF and statins,103,104 both of which may be vasculoprotective. Transgenic mice that carry a TM mutation that reduces its cofactor function in thrombinmediated activation of PC exhibit increased fibrin deposition in the lungs.105 Total inactivation of the TM gene is incompatible with life, and this is likely the same situation for humans, since no cases of homozygous deficiency have been reported. The clinical significance of TM mutations in venous and arterial thrombotic disease in humans otherwise remains controversial. Since the first report of familial thrombophilia associated with a TM gene mutation,106 other mutations and polymorphisms in the coding region and the promoter region have been described, and several have been examined in vitro with mixed results.107 In a large prospective analysis of men, the A455V and -1208-1209TTdelTT variants exhibited a small, but not statistically significant increased risk for coronary heart disease.108,109 In some segments of the population, the Ala25Thr substitution is a risk factor for ischemic heart disease.110 Mutations in the lectin-like domain and in the serine-threonine region have been associated with an increased risk of developing the thrombotic microangiopathy, atypical hemolytic uremic syndrome.111 Overall, genome-wide association studies and advances in proteomics should provide more insights into the relevance of TM polymorphisms in human disease.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


