Rodent Models for the Study of Diabetes
Clayton E. Mathews
Edward H. Leiter
Patients with diabetes share the clinical symptom of hyperglycemia accompanied by either insufficient circulating insulin or inadequate insulin action, but the genetic and environmental interactions leading to the metabolic disorder are complex because of etiopathologic heterogeneity. Because of the multifaceted nature of diabetes, etiologic dissection of diabetes in humans has been difficult. Much of the current understanding of diabetes etiopathogenesis has evolved from analysis of animal models in which either insulin-dependent or insulin-resistant forms of diabetes develop spontaneously. As early as the late 19th century, the importance of animals in the study of human disease was established when Oscar Minkowski (1a,1b) discovered the link between the pancreas and diabetes by pancreatectomizing dogs. The use of dogs and their pancreata by Banting and Best (1c,1d) in the 1920s in their discovery and isolation of insulin as a treatment for humans with insulin-dependent diabetes stands as a landmark in the history of medicine.
While animals have been used for physiologic and pharmacologic studies of diabetes for more than 100 years, animals with genetic syndromes that are models for various forms of diabetes in humans have been developed only during the last 50 years. Since the previous edition of this book, there has been an explosive increase in the number of new mouse models generated by gene-targeting technology wherein specific genes are disrupted (1e). Genetically engineered mutant mice used in diabetes research may or may not serve as good models for the natural course of the development of diabetes in humans but provide valuable tools for evaluating the role of a single gene in the maintenance of either metabolic or immunologic homeostasis (2). The focus of this chapter is rodent models of spontaneously occurring diabetes. We discuss information on targeted mutations of diabetogenic significance only to aid in understanding the spontaneous disease syndrome, especially autoimmune (type 1 or insulin-dependent) diabetes mellitus in the nonobese diabetic (NOD) mouse.
Research involving the available rodent models of both type 1 diabetes and type 2 diabetes (non-insulin-dependent) tends to be compartmentalized, with immunologists and immunogeneticists focusing on the NOD and BB (BioBreeding) rat models of type 1 diabetes. In turn, investigators interested in mechanisms of metabolic control and in obesity focus on the models of type 2 diabetes. This “division of interest” can obscure an important “lesson” taught by viewing the totality of rodent models available. Such a survey would suggest potential pathologic synergism among genomes selected for susceptibility to type 1 or type 2 diabetes. For instance, it is not difficult to envisage that combinations of genes contributing to impaired immune tolerance (a defect associated with type 1 diabetes) might interact deleteriously with genes contributing to impaired β-cell function or impaired glucose tolerance (IGT; both defects proposed to be central to the etiopathogenesis of certain types of type 2 diabetes). One such deleterious interaction was uncovered by mating type 1 diabetes-prone NOD mice to a wild-derived inbred strain carrying a predisposition to obesity (3,4). The biology of the pancreatic β-cell now provides a common meeting ground for researchers in both type 1 and type 2 diabetes. This chapter will describe a variety of relatively new inbred strains generated by sib matings of outbred Swiss (or CD-1) mice. The progenitor-outbred population is considered a model of an outbred human population segregating genetic susceptibility for type 1 and type 2 diabetes. Extensive inbreeding of pairs of mice from this family produced lean, glucose-tolerant strains (SWR, SJL, FVB), strains moderately obese and exhibiting IGT (NON, NSY), and strains developing overt type 2 diabetes with moderate (ALS) or more pronounced obesity (TallyHo) (Table 18.1). An interesting aspect to this family of mice is that it also contains the NOD strain, the most widely used animal model of type 1 diabetes. This chapter will discuss the available models of type 1 and type 2 diabetes in current use. The first portion of the chapter discusses in detail the mouse and rat models of type 1 diabetes. The remainder of the chapter will briefly cover several models of non-immune-mediated insulin-sensitive diabetes and then briefly describe each of the 15 rodent models of type 2 diabetes. The intent of
this chapter is to provide a guide for the interested investigator so that careful selection and use of the appropriate systems can be made.
this chapter is to provide a guide for the interested investigator so that careful selection and use of the appropriate systems can be made.
TABLE 18.1. Characteristics of ICR-Derived Strains | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ANIMAL MODELS OF SPONTANEOUS TYPE 1 (INSULIN-DEPENDENT) DIABETES MELLITUS
Type 1 diabetes mellitus, or insulin-dependent diabetes mellitus (IDDM), is an endocrine disease in humans that, in most cases, results from an autoimmune destruction of the insulin-producing β-cells within the pancreatic islets of Langerhans (5). Clinical presentation of symptoms of type 1 diabetes (fasting hyperglycemia, glycosuria, and ketoacidosis) is usually abrupt in onset and is indicative of destruction of more than 90% of the β-cell population of the endocrine pancreas. The underlying autoimmune processes producing this level of extensive β-cell destruction, however, are believed to be chronic in nature. The two most intensively studied rodent models of autoimmune type 1 diabetes, the NOD mouse and the BB rat, clearly establish that T lymphocytes are both initiators and the final effectors of β-cell destruction (6). Certain clinical and immunopathologic differences distinguish these two models, indicating that the pathways leading to autoimmune type 1 diabetes in humans may be diverse. Because of the greater definition of the mouse genome, coupled with the ease of introducing transgenes and genetically targeted mutations (“knockouts”) into this genome, the NOD mouse has surpassed the BB rat as the more commonly studied model of type 1 diabetes. Yet, given important genera differences between Mus and Rattus in the pathogenesis of type 1 diabetes, investigators considering a rodent model of type 1 diabetes for therapeutic intervention studies should look for efficacy in both the mouse and the rat models if the ultimate goal is to extrapolate such therapies to humans. The clinical heterogeneity of type 1 diabetes is illustrated by a dominant negative mutation in one of the two mouse insulin genes, creating an insulin-dependent syndrome in the absence of autoimmunity in the C57BL/6J- Ins2Akita mouse, a new model to be described below (7). A final example of this clinical heterogeneity is an insulin-responsive diabetes superimposed upon a monogenic obesity syndrome represented in a C57BLKS/J stock congenic for the fat mutation at the carboxypeptidase E locus (Cpefat).
Nonobese Diabetic Mice
ORIGINS AND RELATED STRAINS
NOD is a descriptor string denoting the nonobese diabetic inbred strain developed by inbreeding of ICR (Swiss) mice in Japan (9). The strain is best referred to as NOD, since not all NOD mice will develop diabetes, although most mice of this strain develop insulitis. The discovery of these mice emphasizes the role of serendipity in science. The NOD and certain of the NOD-related inbred strains were developed from Jcl:ICR progenitors at the Shionogi Research Laboratories in Aburahi, Japan, by Dr. S. Makino (8,9,10). Selection for dominant cataract with microphthalmia was the initial objective of the breeding program in Makino’s laboratory. This selective breeding program led to the development of the CTS (cataract Shionogi) strain, in which all mice develop cataracts, and of NCT, a cataract-free control strain. At the sixth generation of inbreeding (F6), Makino noticed that some mice that were free of cataracts exhibited high fasting blood glucose levels. By selective breeding of F6 sibs and their progeny that exhibited this phenotype, Makino hoped to develop a new inbred strain exhibiting spontaneous diabetes. At the same time, he recognized the need for a euglycemic control strain and simultaneously inbred F6 sibs and their progeny that exhibited normal fasting blood glucose levels. In 1974, at the 20th generation of inbreeding (F20), a female spontaneously developed overt type 1 diabetes associated with heavy leukocytic infiltrations within the pancreatic islets (insulitis). Paradoxically, this female was not found in the line being selected for fasting hyperglycemia. Instead, the female with spontaneous type 1 diabetes was found in the line being inbred as a diabetes-free euglycemic control line. The progeny of this diabetic female were the founders of the NOD strain. Since the line originally selected for high fasting glucose levels never progressed to overt diabetes at the time that type 1 diabetes appeared in the euglycemic control (the latter now the NOD) line, the former strain was designated NON, for nonobese normal, and more recently, redesignated as nonobese nondiabetic (8,11). It was serendipitous not only that Makino coselected a euglycemic “control” line but also that his screening of this line was sufficiently rigorous to permit the discovery of that the “exceptional” female and the selection of her progeny to produce the NOD strain. Another serendipitous happenstance was that the husbandry environment in Makino’s vivarium was sufficiently free of murine pathogenic agents such that the diabetic phenotype could develop (see the section “Critical Role of Environment”). It should be noted that while subsequent inbreeding of ICR mice in Japan produced other inbred strains, including the ILI (ICR-L line-Ishibe), NSY (Nagoya-Shibata-Yasuda), ALS (alloxan-induced diabetes susceptible), and ALR (alloxan-induced diabetes resistant), none of the latter have developed spontaneous, autoimmune type 1 diabetes (although some that will be described in sections to follow
have developed IGT, pre-type 2 diabetes, or overt type 2 diabetes) (12,13,14).
have developed IGT, pre-type 2 diabetes, or overt type 2 diabetes) (12,13,14).
Distribution of NOD and its closely related strains was tightly restricted to Japan within the first 5 years of their development (15). However, NOD mice are now widely available from suppliers in Japan (NOD/Shi from Clea, Tokyo), the United States (NOD/LtJ from The Jackson Laboratory, Bar Harbor, ME; and NOD/MrkTac from Taconic, Germantown, NY), and Europe (NOD/HlBom, Bomholtgard, Ay, Denmark). Substrain differences have been reported, notably, the very low incidence of type 1 diabetes present in the NOD/Wehi substrain in Australia (16). To avoid substrain divergence resulting from accumulation of recessive mutations distinct from those being fixed in the source colony, new breeders from the source colony can be imported every 10 to 20 generations to maintain uniformity with the mice being studied by other investigators.
CRITICAL ROLE OF ENVIRONMENT
Investigators studying NOD mice must be aware of the critical role of a specific pathogen-free environment in allowing penetrance of the underlying genetic susceptibility (17). The investigator has to choose between two options: maintaining a NOD breeding colony on site or ordering the requisite number of mice necessary for specific studies. Whichever option is selected, the investigator must house the mice in a specific pathogen-free (SPF) vivarium maintained to high standards of cleanliness and good animal care practice to achieve the diabetes frequencies mentioned above [reviewed in reference (18)]. This is necessary because NOD mice are subject to protective immunomodulation when stimulated by a broad spectrum of extrinsic pathogens, including viruses, bacteria, or their products or when fed semipurified diets lacking diabetogenic catalysts (6).
CLINICAL COURSE OF TYPE 1 DIABETES
In marked contrast to the male gender-biased susceptibility observed in rodent models of type 2 diabetes to be discussed later, one of the mouse-specific characteristics of type 1 diabetes pathogenesis in NOD mice is that it is female gender-biased. This bias is presumably attributable to the development of larger lymphoid organs in females than in males and the stronger response of females to immunization. In high-incidence colonies of NOD mice, the incidence in females typically reaches 80% to 90% by 24 weeks of age, whereas the incidence in males is lower and much more variable, usually reaching more than 40% by 30 weeks of age in SPF colonies. In the author’s (EHL) colony of NOD/Lt males at The Jackson Laboratory, the frequency of type 1 diabetes in males varies from year to year, with a recorded low of 40% by 30 weeks of age and a high of 70% by 24 weeks of age. One of the unusual characteristics of the development of type 1 diabetes in NOD mice is that insulin therapy is not obligatory to sustain life over the first 2 to 4 weeks following the first diagnosis of hyperglycemia or glycosuria. This, in part, represents the ability of diabetic NOD mice to resist development of severe ketoacidosis (19). Diabetic NOD/Lt mice in the author’s colony must exhibit chronically high blood glucose levels (>500 mg/dL) before severe ketonemia can be demonstrated. For example, a diabetic NOD/Lt female with a glucose level of 337 mg/dL had a β-hydroxybutyrate level of only 0.5 mg/dL, a nondiabetic background level. A dipstick test of this mouse’s urine was also negative for ketones (acetoacetic acid). Two chronically diabetic NOD/Lt females (glucose levels of 913 and 954 mg/dL) also were negative for urine ketones but did exhibit blood levels of β-hydroxybutyrate of 10 and 20 mg/dL. Ketone bodies in urine appear only after the establishment of pronounced ketonemia. It should be noted that ketone bodies in the blood of chronically diabetic mice are considerably lower than those in humans, probably because mice can metabolize blood ketones to lactate (20). As hyperglycemia and glycosuria become more severe, mice exhibit polyuria, dehydration, and weight loss. Complete protocols for monitoring the diabetic state have been published elsewhere (18). In vivaria where high levels of NOD intra-islet insulitis develop, but unknown environmental factors suppress the transition to clinical type 1 diabetes, the onset of disease in both males and females can be precipitated by treatment with cyclophosphamide (21). Commonly, 200 mg/kg of body weight is administered intraperitoneally initially, with a second injection given 2 weeks later if required. Cyclophosphamide-accelerated type 1 diabetes is a particularly useful tool for synchronizing activation of diabetogenic effectors in young (7- to 10-week-old) NOD/Lt males.
CONTROL OF BLOOD GLUCOSE LEVELS WITH INSULIN IN DIABETIC MICE
It is exceedingly difficult to maintain stable normoglycemia in a diabetic mouse (NOD or otherwise) by insulin injection, although life span can be extended. A protocol used by Dr. M. Hattori (Joslin Diabetes Center, Boston, MA) (22) entails twice-daily subcutaneous insulin (a 1:1 mixture of regular and NPH insulin administered at 7:30 a.m. and 4:00 p.m.). The dose of insulin (between 0.5 and 1.0 units) is adjusted according to the severity of matinal and evening glycosuria. Table 18.2 shows random glucose and glycosylated hemoglobin (HbA1) measurements in diabetic animals with and without insulin treatment.
TABLE 18.2. Hemoglobin A1 Levels in Diabetic Nonobese Diabetic Mice With and Without Insulin Treatment | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
CONTROL STRAINS FOR NONOBESE DIABETIC MICE
An important issue for investigators studying genes and phenotypes in the NOD mouse is that of an appropriate control. The literature shows that C57BL/6J (B6) or BALB/c are commonly used as “standard” strains against which to compare NOD immunophenotypes or gene sequences. However, since neither strain was Swiss-derived, the differences observed could represent normal variation expected for independently derived mouse strains. Selection of a control strain depends on the nature of the experiment and the hypotheses being tested. A partial listing of possible control strains and their potential uses is provided in Table 18.3. For an investigator interested in associating a particular genetic polymorphism with a phenotype associated with NOD diabetes, it is logical to compare the NOD genotype/phenotype with that found in NOD-related but type 1 diabetes-resistant inbred strains. These include mice with a common Swiss origin but with differences in their major histocompatiblity complex (MHC) haplotype and/or other non-MHC genes (designated Idd loci) conferring various degrees of resistance to type 1 diabetes. Figure 18.1 provides an illustration wherein Swiss-derived strains were surveyed for the phenotype of high percentages of T lymphocytes in the peripheral blood and spleen, an NOD strain characteristic very possibly associated with the resistance of these T lymphocytes to apoptosis (23). This complex phenotype is not unique to NOD; it also is characteristic of the SJL/J strain. SJL/J (H2s) mice are highly susceptible to experimental allergic encephalomyelitis (EAE). A genetic contribution to the SJL strain susceptibility to EAE colocalizes to a 0.15-cM region on chromosome (Chr) 3 that also confers susceptibility to type 1 diabetes (Idd3) in outcrosses of NOD with certain type 1 diabetes-resistant strains (24,25). The logical candidate within this interval is the Il2 gene, with NOD and SJL showing identity for an Il2 gene polymorphism associated with a presumed hyperglycosylated interleukin-2 (IL-2) molecule (26,27). However, it is unknown whether the SJL trait of high numbers of peripheral T cells cosegregates with EAE susceptibility and the SJL Il2 allele. Indeed, SWR/J, another Swiss-derived strain, also shares the Il2 polymorphism with NOD but does not exhibit the T lymphoaccumulation phenotype (Fig. 18.1). This does not exclude the Il2 polymorphism as a contributor to the phenotype but illustrates that genetic control of the trait is oligo- or multigenic. These examples also illustrate the value of comparing NOD to more than one control strain.
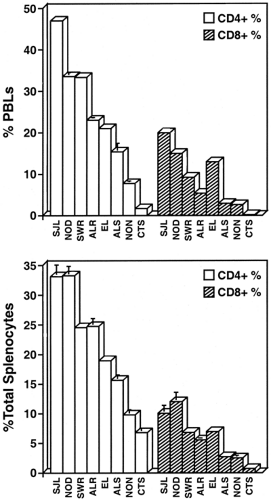 Figure 18.1. Variation in CD4+ and CD8+ T cell percentages of (A) peripheral blood leukocytes (PBLs) and (B) splenic leukocytes from the females of Swiss-derived inbred mouse strains. |
TABLE 18.3. Control Strains and Stocks for Nonobese Diabetic (NOD) Mice | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Because it is H2g7 -identical, but diabetes- and insulitis-resistant, NOR/Lt is an NOD-related strain frequently used to evaluate the effect of changes in non-MHC genes. This is a recombinant congenic strain arising from outcross of NOD with
C57BLKS/J (28). As discussed in detail below, the major genetic determinant of diabetes susceptibility in NOD mice is their H2g7 MHC haplotype (Kd, Ag7, Enull, Db). If an investigator is interested in analyzing the diabetogenic functions of the NOD H2g7 MHC haplotype, stocks congenic for MHC haplotypes conferring resistance to type 1 diabetes are available for analysis of the selection of an autoimmune repertoire. Conversely, the diabetogenic NOD H2g7 haplotype is available for study on the type 1 diabetes-resistant B6 background and in recombinant congenic strains (RCS) between CBA and NOD (29). During the initial period of NOD investigations in Japan, destruction of pancreatic β-cells was firmly established as a T lymphocyte-mediated process [reviewed in reference (10)]. For analysis of the diabetogenic potency of specific NOD T-cell clones or populations of immune effectors, NOD stocks congenic for immunodeficiency-producing mutations blocking development of functional T lymphocytes and/or B lymphocytes are available for adoptive-transfer studies. It has recently been demonstrated that B lymphocytes are essential antigen-presenting cells (APCs) for initiation of type 1 diabetes (B lymphocyte-deficient NOD mice congenic for the Igμ targeted mutation were protected from type 1 diabetes) (30). Therefore, T and B lymphocyte-deficient NOD mice congenic for either the Prkdcscid (severe combined immunodeficiency mutation; henceforth denoted as scid) or targeted mutations in the recombinase-activating gene (Rag1, Rag2) provide useful diabetes- and insulitis-free mice for adoptive-transfer studies without a requirement for prior irradiation (31,32,33,34). The NOD- Rag1 and – Rag2 congenics do not exhibit the DNA repair defects associated with the scid mutation and hence can tolerate sublethal irradiation. Moreover, thymic lymphomagenesis, a strain characteristic of NOD- scid, is slower to develop in NOD- Rag mice (32). Further, these stocks provide an excellent source of NOD pancreatic islets free of insulitic infiltrates. Not included in Table 18.3 is a listing of a growing battery of congenic stocks with defined chromosomal regions bearing non-MHC Idd loci affecting diabetes-related immunophenotypes (35). Certain of these will be discussed below in the section on genetic control of type 1 diabetes.
C57BLKS/J (28). As discussed in detail below, the major genetic determinant of diabetes susceptibility in NOD mice is their H2g7 MHC haplotype (Kd, Ag7, Enull, Db). If an investigator is interested in analyzing the diabetogenic functions of the NOD H2g7 MHC haplotype, stocks congenic for MHC haplotypes conferring resistance to type 1 diabetes are available for analysis of the selection of an autoimmune repertoire. Conversely, the diabetogenic NOD H2g7 haplotype is available for study on the type 1 diabetes-resistant B6 background and in recombinant congenic strains (RCS) between CBA and NOD (29). During the initial period of NOD investigations in Japan, destruction of pancreatic β-cells was firmly established as a T lymphocyte-mediated process [reviewed in reference (10)]. For analysis of the diabetogenic potency of specific NOD T-cell clones or populations of immune effectors, NOD stocks congenic for immunodeficiency-producing mutations blocking development of functional T lymphocytes and/or B lymphocytes are available for adoptive-transfer studies. It has recently been demonstrated that B lymphocytes are essential antigen-presenting cells (APCs) for initiation of type 1 diabetes (B lymphocyte-deficient NOD mice congenic for the Igμ targeted mutation were protected from type 1 diabetes) (30). Therefore, T and B lymphocyte-deficient NOD mice congenic for either the Prkdcscid (severe combined immunodeficiency mutation; henceforth denoted as scid) or targeted mutations in the recombinase-activating gene (Rag1, Rag2) provide useful diabetes- and insulitis-free mice for adoptive-transfer studies without a requirement for prior irradiation (31,32,33,34). The NOD- Rag1 and – Rag2 congenics do not exhibit the DNA repair defects associated with the scid mutation and hence can tolerate sublethal irradiation. Moreover, thymic lymphomagenesis, a strain characteristic of NOD- scid, is slower to develop in NOD- Rag mice (32). Further, these stocks provide an excellent source of NOD pancreatic islets free of insulitic infiltrates. Not included in Table 18.3 is a listing of a growing battery of congenic stocks with defined chromosomal regions bearing non-MHC Idd loci affecting diabetes-related immunophenotypes (35). Certain of these will be discussed below in the section on genetic control of type 1 diabetes.
INSULITIS AND DEVELOPMENT OF TYPE 1 DIABETES
Insulitis, the disruption of islet structure and function by infiltrating leukocytes, is the major pathognomonic feature of the development of type 1 diabetes in NOD mice (8). In these mice, the development of insulitis is not abrupt; rather, the first stages are detectable in females around the time of weaning (4 weeks), at least 2 to 3 months before development of overt diabetes. Islets develop in the perivascular/periductular areas of the pancreas. Insulitis initiates as “periinsulitis,” a pervasive leukocytic infiltrate emanating from the pancreatic vasculature and secretory ducts. Periinsulitis is not unique to NOD/Lt mice; for example, it can be detected in older mice of the related, but diabetes-resistant, NON/Lt strain, as well as in the diabetes-resistant NOR/Lt strain (36,37). However, what distinguishes the histologic appearance of insulitis in NOD mice from that observed either in the T-lymphopenic BBDR (BB diabetes-prone) rat or in humans is the unusually large numbers of leukocytes associated with all the pancreatic vascular spaces, including the lymphatics (lymphoaccumulation) (38). Lymphoaccumulation is not restricted to the pancreas; increased percentages of T lymphocytes are found in peripheral blood, in lymphoid organs, and in other endocrine tissues. Data in Figure 18.1 comparing NOD/Lt to other ICR (Swiss)-derived inbred strains illustrate this phenomenon. Perhaps reflective of this phenomenon, lymphoid tumors are common in aging NOD/Lt mice that remain free of type 1 diabetes (39). Accumulation of leukocytic infiltrates around the perivascular and periductular regions of the pancreas and subsequent development of progressively more severe insulitis (Fig. 18.2), usually initiates between 3 and 5 weeks in NOD females and several weeks later in males. These intrapancreatic lymphoaccumulations primarily, but not exclusively, comprise T lymphocytes (CD4+>CD8+) (40,41,42). B lymphocytes and macrophage/dendritic cells also are found in the infiltrates (43). Although initially few in number, T lymphocytes capable of adoptively transferring type 1 diabetes into T and B lymphocyte-deficient NOD/LtSz- scid recipients can be isolated from islet donors as young as 3 weeks old (44).
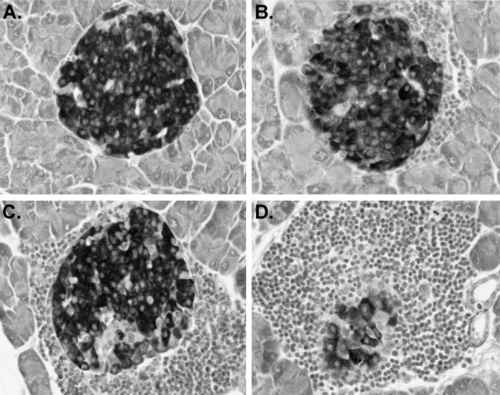 Figure 18.2. Illustration of the various stages of insulitis development in NOD/Lt mice. Islet profiles shown all were photographed in the same pancreas stained with aldehyde fuchsin to identify granulated β-cells. A: Unaffected islet at the level of section examined. B: Islet showing periinsulitis initiating at one pole. C: More extensive insulitis producing erosion of β-cell mass. D: Terminal stages of insulitis. Islet residua eventually comprise exclusively non-β endocrine cells. |
Periinsular leukocytic aggregates around NOD islets usually initiate at one pole but eventually surround the entire islet perimeter (periinsulitis). After initiation of periinsulitis (usually in 3- to 5-week-old females and several weeks later in males), only a subset of islets observed in a microscopic field appears to be penetrated by leukocytes. However, as the mice transit through puberty, an increased prevalence of islet profiles showing intra-islet leukocytic infiltrates, coupled with erosion of between 25% and 90% of the insulin-stainable β-cell mass, is noted (18). The early insulitis, although leading to complete destruction of a percentage of islets in a given pancreas, appears
to be partially compensated by a period of islet growth. During the period between 5 and 12 weeks, NOD/Lt islets are actually quite large in comparison to those of the closely related NON/Lt strain despite the heavy leukocytic aggregations surrounding or penetrating many of the islets in the former. Basal plasma insulin levels are significantly higher in NOD mice of both sexes between 4 and 6 weeks of age compared with levels in C57BL/6 (B6) mice bred in the same vivarium (45). Similarly, NOD fetal pancreas in organ culture secretes higher levels of insulin than does fetal BALB/c pancreas (46). Marked decreases in pancreatic insulin content are demonstrable in NOD/Lt females at around 12 weeks of age and several weeks later in males (47). At this time, increased numbers of atrophic “end-stage” islet profiles are detected. Immunocytochemical staining of these islet residua detects primarily non-β-islet endocrine cells. When more than 90% of the β-cell mass of the pancreas is destroyed, clinical symptoms of diabetes (hyperglycemia, glycosuria, polydipsia, and polyuria) appear abruptly (48). Although intra-islet insulitis initiates early after weaning, some immunoregulatory mechanism apparently keeps many of the infiltrating T lymphocytes in a resting state until a later activation event allows more widespread destruction of the β-cell mass (47,49).
to be partially compensated by a period of islet growth. During the period between 5 and 12 weeks, NOD/Lt islets are actually quite large in comparison to those of the closely related NON/Lt strain despite the heavy leukocytic aggregations surrounding or penetrating many of the islets in the former. Basal plasma insulin levels are significantly higher in NOD mice of both sexes between 4 and 6 weeks of age compared with levels in C57BL/6 (B6) mice bred in the same vivarium (45). Similarly, NOD fetal pancreas in organ culture secretes higher levels of insulin than does fetal BALB/c pancreas (46). Marked decreases in pancreatic insulin content are demonstrable in NOD/Lt females at around 12 weeks of age and several weeks later in males (47). At this time, increased numbers of atrophic “end-stage” islet profiles are detected. Immunocytochemical staining of these islet residua detects primarily non-β-islet endocrine cells. When more than 90% of the β-cell mass of the pancreas is destroyed, clinical symptoms of diabetes (hyperglycemia, glycosuria, polydipsia, and polyuria) appear abruptly (48). Although intra-islet insulitis initiates early after weaning, some immunoregulatory mechanism apparently keeps many of the infiltrating T lymphocytes in a resting state until a later activation event allows more widespread destruction of the β-cell mass (47,49).
CELLULAR AND MOLECULAR ANALYSIS OF THE INSULITIC PROCESS: “MACROPHAGE INSULITIS”
In considering the insulitis process, the reader should be reminded that important substrain and environmental differences distinguish extant colonies of NOD mice around the world (39). Hence, a longitudinal analysis of cellular and molecular events descriptive of insulitic progression in one colony may not exactly match an analysis of the same parameters in a separate colony. This is especially true for reverse transcriptase-polymerase chain reaction (RT-PCR) estimations of cytokine messenger RNAs (mRNAs) expressed in longitudinal studies.
Islet-associated macrophages may mediate cytopathic events indirectly (through secretion of lymphocyte chemoattractants and through the presentation of autoantigens to them) or directly (via secretion of toxic monokines, prostanoids, nitric oxide, and other reactive oxygen species). An immunocytochemical study of NOD mice from a colony in Paris detected ER-MP23+ and MOMA-1+ macrophages/dendritic-like cells as
the first leukocytes to appear around swollen periinsular vessels in 3-week-old mice of both sexes (50); this was termed “macrophage insulitis.” At 7 weeks, periinsular accumulations of leukocytes were reported that contained, in addition to the ER-MP23+ and MOMA-1+ cells, a BM8+ cell described as a phagocytotic macrophage. A shift in location from the periinsular space to the islet interior of BM8+ macrophages was associated with intra-islet insulitis and occurred earlier and at higher frequency in the pancreas of females than in the pancreas of males (50). In NOR/Lt mice, in which heavy periinsulitis develops with age, macrophages penetrate into the islet interior but T cells do not, possibly suggesting absence of a critical chemokine (36). Analysis by RT-PCR of spontaneously developing insulitis in NOD mice shows that, although individual islets within a single pancreas may exhibit a T-lymphocyte population expressing a relatively oligoclonal spectrum of T-cell receptor (TCR) genes, pooled islets exhibit a much more diverse spectrum of TCR clonotypes (51). The islet-reactive CD4+ T-cell clones now available from NOD spleens or islets show a diverse array of TCR α- and β-chain rearrangements (52,53). Interestingly, among the H2-Kd-restricted CD8+ islet-reactive clones isolated from NOD pancreatic islets, a more restricted TCR-α gene utilization has been noted, with up to 30% to 35% using an identical Vα17 to Jα42 TCR gene rearrangement (54,55).
the first leukocytes to appear around swollen periinsular vessels in 3-week-old mice of both sexes (50); this was termed “macrophage insulitis.” At 7 weeks, periinsular accumulations of leukocytes were reported that contained, in addition to the ER-MP23+ and MOMA-1+ cells, a BM8+ cell described as a phagocytotic macrophage. A shift in location from the periinsular space to the islet interior of BM8+ macrophages was associated with intra-islet insulitis and occurred earlier and at higher frequency in the pancreas of females than in the pancreas of males (50). In NOR/Lt mice, in which heavy periinsulitis develops with age, macrophages penetrate into the islet interior but T cells do not, possibly suggesting absence of a critical chemokine (36). Analysis by RT-PCR of spontaneously developing insulitis in NOD mice shows that, although individual islets within a single pancreas may exhibit a T-lymphocyte population expressing a relatively oligoclonal spectrum of T-cell receptor (TCR) genes, pooled islets exhibit a much more diverse spectrum of TCR clonotypes (51). The islet-reactive CD4+ T-cell clones now available from NOD spleens or islets show a diverse array of TCR α- and β-chain rearrangements (52,53). Interestingly, among the H2-Kd-restricted CD8+ islet-reactive clones isolated from NOD pancreatic islets, a more restricted TCR-α gene utilization has been noted, with up to 30% to 35% using an identical Vα17 to Jα42 TCR gene rearrangement (54,55).
As discussed in more detail below, CD4+ T lymphocytes of NOD mice are strongly biased to secrete a T-helper 1 (TH1) spectrum of proinflammatory lymphokines, especially interferon-γ (IFN-γ). IL-12 and IL-18 are two cytokines associated with the deviation of T lymphocytes toward a TH1 cytokine profile. Both are expressed at high levels in NOD splenic and islet-infiltrating macrophages (56,57). In the cyclophosphamide (CY)-accelerated model of diabetogenesis in NOD mice, a TH1-insulitis in NOD/Bom was correlated with upregulation of IL-12 and IL-18 mRNA levels in both spleen and pancreas (56,57). The importance of these monokines is indicated by the observation that onset of type 1 diabetes was retarded by a TH2 deviation achieved by chronic treatment of prediabetic NOD/Lt females with an IL-12 antagonist (58). An elegant quantitative RT-PCR analysis found I-Aβg7 and Igμ transcript levels to be elevated by 20-fold in islets from 20-day-old NOD/Jsd mice compared with nondiabetic control strains (59). However, these markers for intra-islet APCs encompassing both macrophages and B lymphocytes failed to distinguish NOD/Jsd females that transited to overt type 1 diabetes at a high frequency (80% to 85% by 30 weeks) from NOD/Jsd males that were quite resistant to type 1 diabetes (10% to 15% diabetic by 30 weeks). Fox and Danska (36) subsequently used their quantitative RT-PCR techniques to compare a variety of APC-specific and T lymphocyte-specific transcript levels in NOD/Jsd versus the H2g7 identical, but insulitis- and diabetes-resistant NORLt strain. They reported independent genetic regulation of the macrophage insulitis (found in NOD and NOR) from the more advanced phase involving T-lymphocyte penetration (found primarily in NOD) (36). These findings support the concept that clear “checkpoints” in the development of insulitis exist. The swollen periinsular vasculature prior to lymphocyte penetration of islets described above is associated with increased expression of MHC class II and addressin on vascular endothelium. These changes probably facilitate the NOD strain-characteristic T-lymphocyte accumulation in the pancreatic lymphatics and the subsequent extravasation of α4β7-integrin-positive lymphocytes into the periinsular zones (60,61).
CELLULAR ANALYSIS OF THE INSULITIC PROCESS: THE ROLE OF T AND B LYMPHOCYTES
Small numbers of T lymphocytes (both CD4+ and CD8+), as well as B lymphocytes, are present in the early insulitic lesions (41,62). Studies of adoptive transfer into NODLtSz- scid mice confirmed previous reports that pathogenic contributions from both CD4+ and CD8+ subsets are required in the natural progression of the disease (63,64). CD4+ splenic T lymphocytes preactivated in situ in overtly diabetic donors can adoptively transfer type 1 diabetes into NOD/LtSz- scid recipients in the absence of the CD8+ subset. In contrast, both CD4+ and CD8+ subsets are required to transfer type 1 diabetes if isolated from young, prediabetic donors (63). In NOD/LtSz- scid mice, CD8+ T lymphocytes adoptively transferred into NOD- scid recipients cannot home to islets in the absence of CD4+ cells (63). The mutual dependency of CD4+ and C8+ T lymphocytes for initiation of disease in adoptive transfers into NOD- scid is elegantly illustrated by using NOD- scid recipients also homozygous for a targeted β-2 microglobulin (B2m) gene. The islets in these recipients do not express MHC class I cell surface molecules and thus do not serve as CD8+ targets. When splenic T lymphocytes from NOD donors of various ages are transferred into this immunodeficient stock, only those from overtly diabetic donors transfer type 1 diabetes in the absence of class I-expressing islets (54,65). Thus, the earliest initiation, and all but the final phases of autoimmune pancreatic β-cell destruction, require MHC class I-dependent T cells that are able to recognize cognate antigen on MHC class I-expressing β-cell targets. The contributions of CD4+ T cell-secreted cytokines to the development of insulitis through various checkpoints are discussed in the next section.
Adoptive-transfer studies have focused attention on the pathogenic contributions of T lymphocytes, which, when derived from diabetic donors, are able to transfer disease into young recipients without recruitment of host B lymphocytes (66). However, B lymphocytes serving as APCs to amplify T-lymphocyte responses to β-cell autoantigens are essential diabetogenic catalysts in NOD mice. This was established by the discovery that NOD/Lt mice rendered B lymphocyte-deficient either by congenic transfer of a disrupted immunoglobulin heavy-chain gene (Igμ “knockout”) or by treatment with antibody to μ chain rarely developed type 1 diabetes (67,68,69). The absence of B lymphocytes rather than the potential presence of any diabetes resistance genes inadvertently transferred with the genetically disrupted Igμ locus on Chr 12 was demonstrated by abrogation of resistance to type 1 diabetes following repopulation of these mice with B lymphocytes. Autoantibodies, most likely maternal in origin, are found on β-cells from islets of 2- to 3-week-old donors (41). However, such autoantibodies are apparently not required for pathogenesis, since passive transfer of purified serum immunoglobulins from diabetic NOD/Lt donors into these NOD/Lt. Igμnull mice, in contrast to repopulation by B lymphocytes, failed to abrogate the resistance to type 1 diabetes of this stock. It is not known whether the B lymphocytes infiltrating the islet play essential roles in presentation to T lymphocytes of β-cell autoantigens released by T lymphocyte- and/or macrophage-catalyzed β-cell lysis.
MOLECULAR ANALYSIS OF THE INSULITIC PROCESS: THE CHANGING CYTOKINE PROFILE OF ISLET-INFILTRATING LYMPHOCYTES AND THE TH1-TH2 PARADIGM
One of the most unusual aspects of the diabetogenic process in NOD mice is that the autoimmune destruction of β-cells can be drastically retarded by so many different manipulations (70). In essence, the immune dysregulation of NOD mice that predisposes the strain to type 1 diabetes, particularly impaired APC function, can be partially corrected by immunostimulation (71). In some instances, the damage mediated by the insulitic process can be halted or reversed at quite a late stage in disease progression (72). In almost all cases in which the consequences of a protective manipulation have been investigated at the level of
cytokine expression in the pancreas and, more germane, in the islet-infiltrating lymphocytes, a deviation in the spectrum of cytokine profiles expressed is indicated that suggests a TH1→TH2 shift has occurred (73,74). Whereas more than 140 manipulations can drastically retard the onset of hyperglycemia in NOD mice, a much smaller number (mostly entailing transgenic or gene targeting manipulations of genes associated with antigen presentation) are capable of completely suppressing the development of insulitis. This paradox of disease suppression, but not insulitis elimination, has given rise to the concept that a “benign” or “nondestructive” form of insulitis that is present in juvenile NOD mice (or NOD male mice in some colonies) can either be maintained or be elicited through maturity. It is widely held that T lymphocytes producing proinflammatory cytokines associated with the TH1 phenotype, especially IFN-γ, are more likely to effect tissue damage in either spontaneous or experimentally induced T lymphocyte-mediated diseases in mice. Although IL-2 is sometimes used as a paradigm TH1 cytokine, NOD/Lt T lymphocytes are low producers of IL-2 on a per-cell basis, and the NOD’s IL-2 allele may prove to be the susceptibility gene at the Idd3 locus (75). TH2 functions [especially the production of IL-4, IL-10, and transforming growth factor-β (TGF-β)] are viewed as protective, or at least as neutral. The genomic makeup of NOD mice predisposes their CD4+ T lymphocytes to respond to antigenic stimulation in a TH1-biased fashion. As noted previously, NOD macrophages express high levels of IL-12 and IL-18 mRNA (56,57,76,77). This would perhaps partially explain why IFN-γ responses dominate over IL-4/IL-10 secretory responses in NOD mice.
cytokine expression in the pancreas and, more germane, in the islet-infiltrating lymphocytes, a deviation in the spectrum of cytokine profiles expressed is indicated that suggests a TH1→TH2 shift has occurred (73,74). Whereas more than 140 manipulations can drastically retard the onset of hyperglycemia in NOD mice, a much smaller number (mostly entailing transgenic or gene targeting manipulations of genes associated with antigen presentation) are capable of completely suppressing the development of insulitis. This paradox of disease suppression, but not insulitis elimination, has given rise to the concept that a “benign” or “nondestructive” form of insulitis that is present in juvenile NOD mice (or NOD male mice in some colonies) can either be maintained or be elicited through maturity. It is widely held that T lymphocytes producing proinflammatory cytokines associated with the TH1 phenotype, especially IFN-γ, are more likely to effect tissue damage in either spontaneous or experimentally induced T lymphocyte-mediated diseases in mice. Although IL-2 is sometimes used as a paradigm TH1 cytokine, NOD/Lt T lymphocytes are low producers of IL-2 on a per-cell basis, and the NOD’s IL-2 allele may prove to be the susceptibility gene at the Idd3 locus (75). TH2 functions [especially the production of IL-4, IL-10, and transforming growth factor-β (TGF-β)] are viewed as protective, or at least as neutral. The genomic makeup of NOD mice predisposes their CD4+ T lymphocytes to respond to antigenic stimulation in a TH1-biased fashion. As noted previously, NOD macrophages express high levels of IL-12 and IL-18 mRNA (56,57,76,77). This would perhaps partially explain why IFN-γ responses dominate over IL-4/IL-10 secretory responses in NOD mice.
A longitudinal analysis of cytokine gene expression in a colony of NOD/Jsd mice with a marked gender difference in the frequency of type 1 diabetes illustrates the changing cytokine profile that accompanies the progression of NOD mice toward frank hyperglycemia (59). Quantitative RT-PCR analysis showed that islet-infiltrating lymphocytes in males maintained a higher ratio of IL-4/IFN-γ transcripts through puberty than did females, with the increased likelihood of the development of type 1 diabetes in the latter associated with waning expression of TH2 transcripts (59). These types of data reinforce the concept that insulitic destruction of β-cells in NOD mice is represented neither by a continuous linear regression line nor by an abrupt, late-activating process but rather by a gradual and chaotic progression wherein both regulatory events (including the changing endocrine milieu imposed by puberty and reproduction) and stochastic events (the particular subpopulation of CD4+ and CD8+ T lymphocytes “resident” in any given islet) determine the rate at which the diabetes threshold of more than 90% β-cell destruction is reached. The finding that many immunomodulatory interventions that retard type 1 diabetes development in this model produce a TH1→TH2 deviation is generally assumed to be evidence that the deviation itself was the basis for the retarded destruction of β-cells by islet-infiltrating effectors. However, recent analysis of NOD stocks congenic for disrupted genes encoding IFN-γ, IFN-γ receptor β chain, IL-4, and IL-10 has forced a reevaluation of this rather simplistic, but attractive concept.
NOD mice congenic for either a disrupted IFN-γ or IFN-γ receptor β gene (e.g., unresponsive to IFN-γ signaling while producing high levels of TH2 cytokines) develop type 1 diabetes, while NOD stocks homozygous for disrupted IL-4 and IL-10 genes do not become diabetic at an accelerated rate (35,78,79). Although NOD mice congenic for a disrupted IFN-γ receptor α subunit have been reported to be resistant to type 1 diabetes, a subsequent report indicates that the resistance is not the result of the targeted allele on Chr 10 but rather a contribution of strain 129 genome in tight linkage (80,81). Treatment of standard prediabetic NOD mice with the nonspecific immunomodulator BCG (bacille Calmette-Guérin) induced resistance to type 1 diabetes resistance with a TH1→TH2 deviation. This treatment also protected NOD stocks congenic for disrupted IL-4 or IL-10 genes (35,82). Surprisingly, the IFN-γ-deficient stock is not protected by BCG (35). Collectively, these observations suggest that the TH1→TH2 deviation so commonly associated with protective immunomodulatory protocols are consequences, rather than causes, of the deviation. A number of studies have shown that NOD APCs, particularly macrophages, are not fully mature and hence fail to provide normal costimulatory signals such as IL-1, while producing high levels of prostaglandin E2 (83,84). The resistance of NOD peripheral T lymphocytes to activation-induced cell death (AICD) may explain the TH1 bias, since TH1 cells normally are more prone to AICD than are TH2 cells (85,86). Hence, if certain type 1 diabetes-preventative protocols differentially stimulate the AICD of β-cell autoreactive T lymphocytes in a way that preferentially spares those producing TH2 cytokines, it would give the appearance of a TH1→TH2 cytokine shift (35).
MOLECULAR ANALYSIS OF THE INSULITIC PROCESS: THE ROLE OF THE β-CELL AND CANDIDATE β-CELL AUTOANTIGENS
A number of genetic and molecular genetic manipulations designed to limit the T-cell receptor repertoire available to CD4+ and/or CD8+ T cells in the NOD mouse generally fail to prevent type 1 diabetes (87). Hence, the NOD thymus is extremely flexible in its ability to generate a diabetogenic T-cell repertoire, even when rearranged TCR transgenes dictate a repertoire predominantly skewed to an “irrelevant” antigen [e.g., lymphocytic choriomeningitis virus (LCMV) peptide not expressed in NOD β-cells]. The broad spectrum of TCR clonotypes of both CD4+ and CD8+ T cell lines and clones reported to produce β-cytotoxicity effects in the NOD mouse reinforce the diversity of antigens recognized. An unresolved issue is whether a single β-cell autoantigen is targeted initially, followed by “downstream” immune reactivities as other antigens are processed and presented following the initial destruction of a few β-cells. The most commonly discussed candidate autoantigens in humans (based upon spontaneous autoantibody development) are insulin, glutamic acid decarboxylase (GAD), and IA-2, a putative tyrosine phosphatase localized in β-granules (88,89,90,91). Debate about NOD mice has focused on whether insulin or GAD represents the primary autoantigen. T-lymphocyte responses to GAD peptides appear earlier than to other candidate autoantigens, including carboxypeptidase E, peripherin, and heat-shock protein 60 (92). Administration of autoantigens in tolerogenic doses to prediabetic NOD mice can retard the development of type 1 diabetes (93,94). A rather controversial report that GAD67 is the primary autoantigen in NOD β-cells has not been replicated and is suspect because of the claims of easily detectable GAD67 protein in NOD β-cells by immunocytochemistry (95). Based on the plasticity of the T-cell repertoire capable of destroying NOD islets, the most conservative position regarding β-cell autoantigens in the NOD model is that a multiplicity must exist, each individually capable of serving as a target for autoimmune initiation. As noted above, CD8+ T lymphocytes are essential for such initiation, suggesting that the initiating event entails recognition of autoantigenic peptide presented by the β-cell target or vascular endothelium near the β-cells.
Most studies examining the interaction between NOD T lymphocytes and their β-cell targets assume that if the appropriate autoantigen(s) is present and there are T lymphocytes in the periphery with the appropriate TCR clonotype(s), then type
1 diabetes will ensue. This is not necessarily true. Both rodent and human β-cells have very weak defenses against oxidative stress (96). NOD β-cells appear typical of most mouse strains in being quite sensitive to cell death mediated by combinations of monokines and IFN-γ. Analysis of β-cells from ALR/Lt mice, a strain closely related to NOD and selected for resistance to type 1 diabetes induced by a low dose of alloxan, has demonstrated that genes expressed at the β-cell level, and presumably associated with dissipation of reactive oxygen species, can protect against both toxic combinations of cytokines and monokines, as well as against islet-reactive cytotoxic T cells isolated from NOD islets and capable of recognizing antigens presented by ALR/Lt islets (13,97,98,99).
1 diabetes will ensue. This is not necessarily true. Both rodent and human β-cells have very weak defenses against oxidative stress (96). NOD β-cells appear typical of most mouse strains in being quite sensitive to cell death mediated by combinations of monokines and IFN-γ. Analysis of β-cells from ALR/Lt mice, a strain closely related to NOD and selected for resistance to type 1 diabetes induced by a low dose of alloxan, has demonstrated that genes expressed at the β-cell level, and presumably associated with dissipation of reactive oxygen species, can protect against both toxic combinations of cytokines and monokines, as well as against islet-reactive cytotoxic T cells isolated from NOD islets and capable of recognizing antigens presented by ALR/Lt islets (13,97,98,99).
GENETICS OF TYPE 1 DIABETES
“Idd” loci: What and How Many?
A provisional nomenclature exists to describe murine chromosomal regions carrying genes capable of modulating susceptibility of type 1 diabetes. Such loci, designated “Idd” loci are uncovered either by out-crossing NOD mice to type 1 diabetes-resistant strains of mice and then doing segregation analysis or by genetically disrupting specific genes and analyzing the consequences on the development of type 1 diabetes (100). Segregation analysis has shown that many strains harbor potential “Idd” susceptibility contributions but that the NOD strain is unfortunate in terms of the large numbers randomly inbred into them during the generation of the strain. If the large numbers of genes shown by gene-targeting experiments to be essential for mediation of the diabetogenic process are excluded and only “Idd” loci identified by out-crossing NOD to type 1 diabetes-resistant strains are considered, linkage of more than 19 loci on 10 chromosomes have been reported [reviewed in references (39,87)]. Although originally given sequential numbers as they were reported in the literature, segments of chromosomes now carrying genes capable of modifying the frequency of type 1 diabetes are simply being described by molecular markers physically delimiting the region (generally simple sequence repeat-containing alleles). As noted above, certain of the susceptibility linkages derive from the genomes of type 1 diabetes-free outcross partner strains, and in one case, admixture of a wild-derived strain’s genome produced a male gender-biased type 2 diabetes (3,4). To further complicate matters, a chromosomal segment identified initially through segregation analysis as containing a single “Idd” generally yields multiple loci when subcongenic analysis is conducted. Classical genetic segregation analysis greatly underestimates the actual number of genes required for the development of type 1 diabetes since transgenic and gene-targeting technologies being applied to analyze the genetic control of the development of type 1 diabetes in NOD mice are providing an ever-expanding panoply of contributory “Idd” loci, only a few of which will be discussed. Although most genomic manipulation by transgenic or gene “knockout” technology has focused on genes expressed in the immune system, it is now clear that genes expressed systemically and within the pancreatic β-cell can also be important modifiers of susceptibility (101). A discussion of the MHC-linked diabetogenic contributions (“Idd1”) will suffice to underscore the complexity of “Idd” loci and their interactions.
Paramount Role of Major Histocompatiblity Complex
The first genetic analysis of type 1 diabetes in the NOD mice reported a recessive susceptibility for clinical disease linked to MHC (102). Because the MHC complex contains genes essential for antigen processing and presentation, as well as for immune cell function, and given evidence indicative of broad-based immunotolerogenic defects in NOD mice, it is not surprising that H2g7, encompassing both common class I alleles, a rare class II Ag7 allele, and absence of class II E (the HLA-DR homologue), provides the major component of genetic susceptibility (103,104,105). Indeed, specific amino acid substitutions in the H2-Abg7 chain reflect similar residues present in the human HLA-DQ0302 allele linked to increased susceptibility to type 1 diabetes in whites. These include a histidine residue at position 56 and a serine residue at position 57 of the β chain. Recent radiographic crystallographic analysis helps to understand the diabetogenic properties of the I-Ag7 molecule. The NOD I-Ag7 molecule behaved very much like I-Ad, a MHC class II molecule that shares the same β chain with I-Ag7 but contains proline and aspartic acid at residues 56–57 of the β chain and protects from diabetes. Binding affinities of a large collection of peptides are comparable for I-Ag7 and I-Ad. Presence of negatively charged residues in the C-terminal region of the peptide increases binding affinity to I-Ag7. Exchange of the aspartic acid, as found in I-Ad, by a serine at position β57, increases the size of the P9 pocket of I-Ag7 and exposes positively charged residues. As a consequence, the I-Ag7 P9 pocket can bind negatively charged residues and residues with larger side chains than does I-Ad. These two features expand the peptide repertoire of I-Ag7 by several-fold in comparison to I-Ad. Immunoprecipitation studies suggested that I-Ag7-peptide complexes are more unstable than similar MHC class II complexes from B6 (106). This might imply that I-Ag7 bound more peptides, but with lower affinities such that cytopathic T-effectors are not centrally or peripherally deleted.
Molecular Dissection of “Idd1”
The diabetogenic relevance of the nested set of five nucleotide substitutions between position 248–252 converting a conserved proline residue at amino acid position 56 to histidine and aspartic acid 57 to serine was confirmed separately by production of transgenic stocks of NOD mice expressing Abg7 alleles modified by site-specific mutagenesis to convert either the histidine 56 residue to proline or the serine 57 residue to aspartic acid (107,108,109,110). Further demonstration that “Idd1” comprised at least two linked, but separable susceptibility components was provided by the demonstration that the development of type 1 diabetes was also drastically suppressed or completely prevented in stocks of NOD mice expressing H2-Ea transgenes that restored H2-E expression on APC (107,109,110,111). It is not only the MHC class II region that contributes susceptibility in NOD mice but rather multiple loci in the extended haplotype. The requirement for MHC class I expression for cytopathic CD8+ T-lymphocyte targeting of islet β-cells has been detailed above. It is noteworthy that extended haplotype analysis of human genes in linkage disequilibrium with HLA-DR and – DQ alleles conferring susceptibility to type 1 diabetes indicates additional susceptibility components toward the class I region (112). Analysis of NOD stocks congenic for recombinant haplotypes generated by crossover between H2g7 and H2209 provide evidence of additional “Idd” contributors centromeric to a Lmp2 recombinatorial hotspot that both includes and extends proximal to the H2-K gene (113). One of these could represent “Idd16,” initially mapped in linkage disequilibrium to Idd1 on Chr 17 (114). Because of this complexity, it is difficult to discuss whether the haplotype contributes susceptibility in a dominant or recessive fashion. Segregants heterozygous for H2g7 may develop subclinical levels of insulitis (suggesting dominant H2g7 contributions to this important diabetes subphenotype) and may even develop overt type 1 diabetes following cyclophosphamide treatment (115,116). However, type 1 diabetes rarely develops spontaneously in such heterozygotes, especially when an Ea
product is expressed by the other H2 haplotype (116). In this regard, the NOD model diverges from the human situation, where certain HLA heterozygous haplotypes increase rather than suppress susceptibility to type 1 diabetes (117). In summary, “Idd1” is a collective descriptor for multiple disease-predisposing alleles within or linked to the MHC complex.
product is expressed by the other H2 haplotype (116). In this regard, the NOD model diverges from the human situation, where certain HLA heterozygous haplotypes increase rather than suppress susceptibility to type 1 diabetes (117). In summary, “Idd1” is a collective descriptor for multiple disease-predisposing alleles within or linked to the MHC complex.
Diabetogenesis Entails Complex Major Histocompatibility Complex-Non-major-Histocompatibility Complex Interactions
Susceptibility to type 1 diabetes in segregating hybrids following outcross of NOD to other mouse strains is inherited as a polygenic threshold liability, requiring a complex interaction between the H2g7 haplotype and numerous other MHC-unlinked genes (118). None of the non-MHC genes present in the NOD genome contributes the same risk as the extended MHC haplotype, but homozygosity for H2g7 in the absence of a diverse collection of the NOD strain’s collection of non-MHC susceptibility modifiers is insufficient to trigger type 1 diabetes (119). Unraveling the genetic basis for the development of type 1 diabetes in the NOD mouse is complicated by interactions between the NOD mouse’s diabetogenic H2g7 and the welter of non-MHC “Idd” loci, none of which alone is sufficient to elicit type 1 diabetes. This can be demonstrated by developing B6 stocks congenic for long “Idd” susceptibility intervals from NOD (120). Although some level of periinsulitis can be seen, severe insulitis does not develop, even when bicongenic stocks are analyzed that carry both the non-MHC interval and the NOD “Idd1” genes on Chr 17 (121). Genetic analysis is further complicated by the finding that the set of non-MHC genes segregated are not constant but vary depending upon the type 1 diabetes-resistant partner strain used in the outcross. An “Idd” exerting a strong effect in one cross may not be detectable at all in another outcross combination. The situation is made even more complex by the knowledge that strong environmental influences, as well as intergenic interactions, affect gene penetrances and ultimate presentation of clinical disease. The interested reader is referred to a recent review of current knowledge of the locations and potential functions of the most intensively studied of the known non-MHC loci (87). Several examples are provided below to illustrate the nature and complexity of the intergenic “cross-talk” required for diabetogenesis.
Illustrations of Diabetogenic Epistasis
Segregation analysis between H2g7 -identical NOD/Lt and NOR/Lt mice permitted elucidation of a Chr 2/Chr 17 (MHC) interaction. Even though the MHC class I alleles of NOD and NOR mice (H2Kd, H2Db) are commonly expressed in non-autoimmune-prone strains, they acquire diabetogenic function in NOD mice (122). A relatively long segment of NOR/Lt-derived genome on Chr 2 containing at least two resistance genes (collectively described as “Idd13”) provided partial protection from type 1 diabetes in a NOD congenic stock (123). Included in this congenic interval was an NOR-derived B2mb allele, replacing the equally common B2ma allele expressed by NOD. This seemed an unlikely candidate gene for an “Idd13” component since these two MHC class I-binding proteins differed at only a single amino acid and did not alter total expression levels of MHC class I molecules. However, conformational analysis indeed showed that dimerization of NOD MHC class I Kd and Db chains with the alternative β2m isoforms differentially altered their structural conformation (123). The type 1 diabetes-free NOD. B2mnull stock described above provided the means for a rigorous test of B2m candidacy by transgenic insertion directly into these zygotes of transgenes encoding either β2m isoform. A CD8+ T-cell repertoire was positively selected in both lines, but only the line with the reconstituted expression of the B2ma (NOD-type) isoform developed type 1 diabetes (87). This represents the first empirical demonstration of the molecular nature of a non-MHC “Idd” candidate.
In addition to this Chr 2/Chr 17 (MHC) epistatic interaction, a recent genetic analysis of the markedly different course of insulitis progression in NOR versus NOD has suggested an epistatic interaction between the Chr 2 “Idd13” region containing the B2m candidate and Chr 1 (“Idd5.2” region) (124). Epistasis has also been reported among “Idd” loci on Chr 3, specifically the Idd3 locus marked by the Il2 allele and more distal loci denoted as “Idd10/17/18” (116).
Genetics of Mouse Type 1 Diabetes and the Identification of Human Type 1 Diabetes Genes
Although the H2g7 haplotype was originally thought to be unique to the NOD mouse, this haplotype is shared by the type 1 diabetes-free ILI strain (125). Several of its diabetogenic components (e.g., the class II alleles and the H2-Kd allele) also present in the related ALR and CTS strains (126,127). Since the ALR/Lt strain exhibits remarkable genetic resistance to autoimmune stress, it is clear that “diabetogenic” MHC alleles are not inherently diabetogenic but acquire diabetogenic potency only in the context of the non-MHC background (101). Moreover, not all of the susceptibility alleles will derive from the NOD genome, and some admixtures can produce type 2 rather than type 1 diabetes (4). What is the implication of this complexity for the inheritance of type 1 diabetes in humans? Will identification of mouse “Idd” loci identify possible homologous susceptibility regions in the human genome? There are a number of reasons why many of the known non-MHC mouse “Idd” loci are not matched by type 1 diabetes loci in the syntenic regions of the human genome. Important genus-specific differences distinguish mice from humans. The human IDDM2 locus has been identified in both association and linkage studies and is defined by polymorphisms in a VNTR (variable number of tandem repeats) upstream of the human insulin (INS) gene (128). Although proinsulin/insulin represents a major type 1 diabetes autoantigen in both humans and NOD mice, an NOD homologue of the human INS gene (mouse Ins2 on distal Chr 7) has not been detected in most segregation analyses (129,130). This lack of concordance might be expected because mice express two genes for insulin (the additional gene is Ins1 on Chr 19). The “Idd3” (Il2) linkage is one of the strongest and most commonly identified non-MHC loci segregating in NOD outcrosses; the IL2 gene in the homologous 4q26-q27 region in humans has not yet been implicated as a major type 1 diabetes-susceptibility locus (25). Of all the non-MHC loci identified in NOD outcrosses, the “Idd5.1” region containing the Cd152 (Ctla4) locus may be reflected by the IDDM12 linkage to the homologous region on human 2q33 (131,132). Despite the finding that non-MHC genes contributing to the development of type 1 diabetes in NOD mice often are not reflected by demonstration in humans of linkage to the syntenic region, the value of the mouse system is to elucidate specific immunologic or endocrinologic dysfunctions that are entrained when sufficient numbers of “Idd” genes are present. While the human IL2 structural gene has not been implicated as a gene for type 1 diabetes, it is certainly possible that an unlinked gene, acting in trans, could confer a higher risk for type 1 diabetes by affecting IL-2 glycosylation state, as is implicated in the NOD mouse. Even if the controlling genes are not transposable from mice to humans in a one-to-one ratio, many of the phenotypes diagnostic of high susceptibility may be common. Further discussion of the similarities and differences between humans and mice in this regard is found in Chapter 21.
CELLULAR BASIS FOR GENETIC SUSCEPTIBILITY
Defective Antigen-Presenting Cell Functions
Although T lymphocytes often are the focus of studies describing immunotolerogenic defects in the NOD mouse, studies using bone marrow chimeras have demonstrated that the origin of many of the defects can be traced to APC dysfunction (macrophage, dendritic cells, and B lymphocytes). Diminished levels of antigen presentation by MHC molecules represent one likely mechanism whereby immune tolerance is not fully acquired. Both intrathymic and peripheral tolerance induction also requires costimulatory signals delivered to T lymphocytes by APC. Considerable evidence indicates that the APC of NOD mice maintained in SPF environments are deficient in ability to provide sufficiently high levels of costimulation. NOD macrophages maturing from bone marrow precursors do not become as functionally mature as macrophages from control strains (83,133,134). This blunted functional development is indicated by subnormal lipopolysaccharide (LPS)-stimulated IL-1 secretion and by reduced CD86 (B7-2) gene expression (83,133,135). D-galactosamine-sensitized NOD mice are resistant to doses of LPS and TNF-α that produce lethal hepatocellular injury and apoptosis in B6 mice (136). These hypofunctions are negatively reinforced by constitutive expression of the normally inducible prostaglandin E2 synthase gene (Pgst2 on Chr 1) and hence secrete higher than normal levels of prostaglandin E2 (84). Defects in both the high-affinity Fcγ1 receptor and Fcγ2 receptor have been reported (84,137). Both defects might be expected to affect the ability of macrophages to phagocytose monomeric IgG2c and IgG2b antibodies, respectively (the NOD mouse expresses the IgG2c and not the IgG2a isotype as is commonly assumed) (138). Glutathione levels are lower in NOD macrophages than in the NOR/Lt control strain, possibly contributing both to reduced capacity to process and present MHC class II-restricted antigens to CD4+ T cells and to the TH1 bias in response to antigen priming (139,140). Some anomalies in development of marrow-derived NOD dendritic cells have also been reported (141). Vitamin D metabolism is also disturbed in NOD macrophages (141a). The ability of many different microbial immunomodulators, such as CFA, BCG, and OK432, to upregulate APC function and to promote T-lymphocyte apoptosis links APC defects in costimulatory signaling to this major defect in the NOD T-lymphocyte compartment (e.g., their resistance to multiple forms of apoptosis) (35,142). Indeed, the high production of prostaglandin E2 from NOD macrophages could contribute to T-lymphocyte apoptosis resistance by antagonizing TCR-coupled protein kinase C (PKC) second-messenger activities.
Thymocyte/T-lymphocyte Anomalies
The T-lymphocyte accumulation characteristic of NOD mice, reflected at its most extreme by the formation of what appear to be lymphoid follicles in the perivascular spaces of the pancreas and submandibular salivary glands, is a secondary reflection of resistance to AICD. Both NOD thymocytes and peripheral T lymphocytes are relatively resistant to AICD, as well as to apoptosis induced by dexamethasone, irradiation, or stress (143,144,145,146,147,148). A less severe resistance of male thymocytes to AICD may explain the reduced male susceptibility to type 1 diabetes (149). As discussed above, elevated prostaglandin E2 levels may account for the association between poor proliferation in response to TCR cross-linking agents and reduced ability of thymocytes/T lymphocytes to activate TCR-coupled PKC second-messenger pathways (150). The earliest thymic immigrants, especially in females, may be enriched for effector T lymphocytes versus regulatory cells, because thymectomy at 3 weeks of age accelerates the onset of type 1 diabetes in females, but interestingly, not in NOD males (151).
IL-2 is now recognized not only as a growth factor for T lymphocytes but also as a required factor for AICD. The Il2b allele expressed by NOD is distinguished from the Il2a allele expressed by NON, BALB/c, and C57BL strains by presence of a T→C point mutation producing at residue 6 a Ser→Pro substitution, a four-amino-acid insertion at residue 8 (Ser-Ser-Pro-Thr), and a deletion of four glutamine residues within a polyglutamine stretch between residues 19 and 30 (152,153). Il2 is an attractive candidate gene not only because IL-2 treatment of prediabetic NOD mice suppresses the development of type 1 diabetes but also because genetic disruption of the Il2 gene produces severe lymphoproliferative disease with widespread tissue infiltrates, including pancreatitis and insulitis (71,154). Production by NOD splenic T lymphocytes of bioactive IL-2 on a “per-cell” basis was less than that from SWR/Bm, another Swiss-derived strain that shares the Il2b allotype (155,156). The NOD-produced molecule is associated with a higher glycosylation state, and this may affect either half-life or ability to induce AICD (157).
Numerous defects in T-regulatory cell functions have been reported in NOD mice (158). Among the presumed regulatory cells are natural killer T (NKT) cells. Representing a small population (<3%) of peripheral lymphoid cells, they are CD3+, CD8–, and either CD4– or CD4+. Their selection entails recognition of glycolipid antigens presented by the nonclassical MHC class I-like CD1 molecule (159). Deficiencies in both number and immunoregulatory function of NOD NKT cells have been reported (160,161). These deficiencies include a low level of IL-4 secretion, a defect that can be corrected either by IL-7 treatment or by congenic transfer of B6 genes regulating natural killer (NK) and NKT function (including their NK1.1 phenotypic marker) in the “Idd6” type 1 diabetes-resistance region (162,163,164). Enriched populations of cells with an NKT phenotype can protect NOD mice from type 1 diabetes, leading to the suggestion that TH2 cytokine secretion by these cells mediates the protection (162). However, NKT cells stimulated in vitro and in vivo actually produce more IFN-γ than IL-4 (165,166). Indeed, type 1 diabetes-resistant NOD.B6-“Idd6” mice produce more serum IFN-γ upon stimulation with α-galactosyl ceramide than do standard NOD mice (164). Hence, the molecular basis for NKT-mediated protection may be more complicated than a simple TH1 to TH2 deviation.
POLYGLANDULAR AUTOIMMUNITY
Salivary and Lacrimal/Harderian Glands
The multiplicity of immunodeficiencies characteristic of NOD mice leads to leukocytic infiltrations into endocrine organs in addition to pancreatic islets. Particularly notable are heavy infiltrations of the submandibular salivary gland (sialoadenitis) and the lacrimal and harderian gland of the eye (dacryoadenitis). Both sialoadenitis and dacryoadenitis in NOD mice develop after puberty, considerably later than insulitis. Splenic T lymphocytes from both overtly diabetic and young prediabetic NOD mice adoptively transfer these lesions into NOD/LtSz- scid mice (167). Although T lymphocytes are present, the infiltrates are not associated with a loss of glandular acinar cells as extensive as the progressive loss of β-cells in the pancreas (168,169). Since the sialoadenitis impairs salivary flow rates and protein content, and since NOD mice also develop leukocytic infiltrates in their lacrimal and harderian glands, NOD mice may serve as a model for Sjögren syndrome in humans (170). Although loss of salivary gland structural integrity is not as extensive as in Sjögren syndrome in humans, xerostomia (“dry
mouth”), a clinical condition often associated with both Sjögren syndrome and type 1 diabetes in humans, is reflected in NOD mice (170). The dacryoadenitis develops at fourfold higher frequency in postpubertal males than females and can be suppressed by male castration, while tamoxifen treatment significantly increased frequency of this lesion in males (171). Its autoimmune nature was documented by upregulation of inflammatory TH1 cytokine genes in the infiltrating T lymphocytes (171). As is the case for insulitis, salivary gland infiltrates are composed primarily of CD4+ cells with smaller numbers of CD8+ cells and with a diversity of TCR clonotypes represented (172). Insulin and insulin-like growth factors I and II are found at low concentrations in mouse saliva, as well as in parotid and submandibular glands, and hence may represent a potential target antigen (173). TNF-α is apparently involved in lesion development, since treatment of NOD mice with a soluble TNF-receptor p55 subunit inhibits infiltration in both submandibular and lacrimal glands (174). NOD strain-specific anomalies in the development of the submandibular gland were found in NOD/LtSz- scid mice lacking functional T and B lymphocytes (175). Hence, these developmental anomalies may trigger sialoadenitis when a functional immune system is present. An interesting aspect of the sialoadenitis in NOD mice is the potential role of autoantibodies. B lymphocyte-deficient NOD/Lt. Igμnull mice rarely develop type 1 diabetes, but they do develop sialoadenitis and dacryoadenitis in the absence of insulitis and type 1 diabetes (176). Passive transfer into these immunoglobulin-deficient mice of serum immunoglobulins either from standard NOD mice or from patients with primary Sjögren syndrome, but not from healthy controls, inhibited binding of ligands to muscarinic receptors on salivary membranes (176).
mouth”), a clinical condition often associated with both Sjögren syndrome and type 1 diabetes in humans, is reflected in NOD mice (170). The dacryoadenitis develops at fourfold higher frequency in postpubertal males than females and can be suppressed by male castration, while tamoxifen treatment significantly increased frequency of this lesion in males (171). Its autoimmune nature was documented by upregulation of inflammatory TH1 cytokine genes in the infiltrating T lymphocytes (171). As is the case for insulitis, salivary gland infiltrates are composed primarily of CD4+ cells with smaller numbers of CD8+ cells and with a diversity of TCR clonotypes represented (172). Insulin and insulin-like growth factors I and II are found at low concentrations in mouse saliva, as well as in parotid and submandibular glands, and hence may represent a potential target antigen (173). TNF-α is apparently involved in lesion development, since treatment of NOD mice with a soluble TNF-receptor p55 subunit inhibits infiltration in both submandibular and lacrimal glands (174). NOD strain-specific anomalies in the development of the submandibular gland were found in NOD/LtSz- scid mice lacking functional T and B lymphocytes (175). Hence, these developmental anomalies may trigger sialoadenitis when a functional immune system is present. An interesting aspect of the sialoadenitis in NOD mice is the potential role of autoantibodies. B lymphocyte-deficient NOD/Lt. Igμnull mice rarely develop type 1 diabetes, but they do develop sialoadenitis and dacryoadenitis in the absence of insulitis and type 1 diabetes (176). Passive transfer into these immunoglobulin-deficient mice of serum immunoglobulins either from standard NOD mice or from patients with primary Sjögren syndrome, but not from healthy controls, inhibited binding of ligands to muscarinic receptors on salivary membranes (176).
Thyroid Glands
Another common histopathologic lesion described in NOD mice is the presence of leukocytic infiltrates in the thyroid follicles (thyroiditis). This is of interest because autoimmune thyroid disease (Grave disease or Hashimoto thyroiditis) is frequently associated with type 1 diabetes in humans. However, the spontaneously developing lesions in NOD are not so severe that clinical manifestations of hypothyroidism appear. If goiters are induced and followed by iodine loading, severe lesions causing hypothyroidism are elicited (177,178).
Splenocytes from diabetic NOD/Lt donors also adoptively transferred thyroiditis into NOD/LtSz- scid recipients, albeit at a lower efficiency than insulitis (167). Spontaneous development of thyroiditis varies widely among different NOD colonies, with differences in environmental factors such as dietary iodine content representing possible explanations (179). Parathyroiditis has also been reported in one colony (180). Criteria for assessing thyroiditis may be partially responsible for the variability between colonies. A confounding factor in the diagnosis of both thyroiditis or parathyroiditis in NOD mice is the presence of a high frequency of thymic ectopies to the thyroid gland (181). Thymic ectopies are present at ∼20% frequency in this author’s (EHL) NOD/Lt colony, but significant leukocytic infiltration into thyroid interstitium or into follicles is relatively rare (∼5% of mice sampled to 16 weeks of age). In a colony of NOD mice with a low incidence of diabetes accompanied by a high incidence (77%) of subclinical thyroiditis, as well as by antithyroid autoantibodies in older mice, thyroiditis was clearly independent from spontaneous development of type 1 diabetes (182). This was reinforced by the findings in a NOD colony in Paris with a high frequency of type 1 diabetes accompanied by a high incidence (20.5%) of thyroiditis in nondiabetic survivors (mostly males) aged to 90 weeks (183). When this incidence was compared with that of a diabetes-resistant NOD stock congenic for the H2k haplotype (associated with high susceptibility of the CBA/J inbred strain to experimentally induced thyroiditis), a significantly higher (30.3%) frequency of thyroiditis was noted (183). An age-associated increase in MHC class II expression on macrophages and thyroid epithelial cells has been noted (184). Clinically significant lesions comparable to Hashimoto thyroiditis can be induced in NOD mice by administration of a diet high in sodium iodide following induction of goiters using a diet low in iodine in combination with treatment with phenylthiourea (178). NOD/Mrk congenic for an intra-MHC recombinant haplotype (H2h4) expressing H2-Kk and H2-Ak alleles will develop thyroiditis following exposure to sodium iodide alone (116).
AGING-ASSOCIATED PATHOLOGIES
Cancer
In addition to these polyendocrinopathies, leukocytic infiltrates of varying intensity can be found in liver (mild focal hepatitis), kidney (focal nephritis), and muscle (myositis) (185). If development of type 1 diabetes in NOD mice is prevented such that life span is extended, this strain exhibits a virtual “Pandora’s box” of pathologies (185). A spectrum of tumors has been identified in the research colony at The Jackson Laboratory. Of these, lymphomas are those diagnosed most frequently. This propensity for lymphomagenesis in the absence of diabetes is starkly illustrated by the near 100% frequency of thymic lymphomas in NOD/LtSz- scid mice and B (follicle center) cell lymphomas in NOD/LtSz- Ragnull congenic mice (32,186). Osteosarcomas, a relatively rare tumor in mice, are also detected at a relatively high frequency in standard NOD/Lt mice in The Jackson Laboratory research colony (186).
Hearing Defects
NOD mice are severely hearing-impaired, developing a progressive hearing loss as assessed by auditory-evoked brain-stem responses (187,188). In most NOD mice, hearing loss progresses to complete deafness by 3 months. CBA/J mice have normal hearing. Three recombinant congenic stocks have been produced by outcross of NOD mice to CBA/LtJ, followed by a backcross to CBA and then inbreeding of first backcross progeny (29). Testing of auditory-evoked brain-stem response of these three recombinant congenic lines carrying mixtures of NODLt and CBALtJ genes indicates that this trait is under polygenic control, since one line hears as well as CBA and two lines have moderately impaired hearing. Because all three lines share the H2g7 MHC haplotype with NOD, genes within this complex are excluded.
Hemolytic Anemia
Hemolytic anemia accompanied by development of Coombs-positive autoantibodies, reticulocytosis, splenomegaly, and jaundice, has been observed in aging (older than 200 days) mice of both the NOD/Wehi and NOD/Lt substrains maintained in Australia (189). In the low-diabetes-incidence NOD/Wehi substrain, 14 of 17 nondiabetic mice were affected by 550 days.
EXPERIMENTALLY INDUCED LESIONS
NOD mice are susceptible to a variety of experimentally induced organ-specific immune diseases, including drug-induced diabetes, lupus, colitis, encephalomyelitis, and thyroiditis. Not surprisingly, young NOD/Lt males, like outbred CD1 males, are susceptible to induction of type 1 diabetes by multiple low doses of the fungal antibiotic streptozotocin (190). This sensitivity is independent of autoimmune T-cell involvement,
since NOD/LtSz- scid males are even more sensitive to low doses of this diabetogen (190). Injection of a single dose of 2.6 × 107 heat-killed Mycobacterium tuberculosis (BCG) intravenously into 8-week-old NOD mice prevented diabetes but precipitated a syndrome similar to systemic lupus erythematosus (191). Exposure of NOD/Lt mice to 3.5% dextran sodium sulfate in the drinking water for 5 days elicits a severe colitis (192). Experimental allergic encephalomyelitis can be induced in NOD mice using a peptide fragment (residues 56–70) in adjuvant from proteolipid protein (193). The NOD allele at the “Idd3” locus (in the region of the gene encoding IL-2) has been shown to be a component of this strain’s high susceptibility to encephalitis induction (24). A similar demyelinating disease can be induced by injection of the MOG peptide (residues 35–55) in adjuvant; this model is prevented by treatment with anti-IL-12 antibody. A single intravenous injection of pertussin without adjuvant into NOD mice induced a T-cell-mediated encephalitis (194).
since NOD/LtSz- scid males are even more sensitive to low doses of this diabetogen (190). Injection of a single dose of 2.6 × 107 heat-killed Mycobacterium tuberculosis (BCG) intravenously into 8-week-old NOD mice prevented diabetes but precipitated a syndrome similar to systemic lupus erythematosus (191). Exposure of NOD/Lt mice to 3.5% dextran sodium sulfate in the drinking water for 5 days elicits a severe colitis (192). Experimental allergic encephalomyelitis can be induced in NOD mice using a peptide fragment (residues 56–70) in adjuvant from proteolipid protein (193). The NOD allele at the “Idd3” locus (in the region of the gene encoding IL-2) has been shown to be a component of this strain’s high susceptibility to encephalitis induction (24). A similar demyelinating disease can be induced by injection of the MOG peptide (residues 35–55) in adjuvant; this model is prevented by treatment with anti-IL-12 antibody. A single intravenous injection of pertussin without adjuvant into NOD mice induced a T-cell-mediated encephalitis (194).
The BioBreeding Rat
INSULITIS AND DEVELOPMENT OF TYPE 1 DIABETES: COMPARISONS WITH NONOBESE DIABETIC
BioBreeding (BB) rats are a diabetes-prone strain derived from outbred Wistar rats from a commercial colony in Canada (195). The interested reader is referred to comprehensive recent reviews (196,197,198,199,200). Spontaneous type 1 diabetes was discovered in 1974, and selective breeding for that phenotype ensued. As with NOD mice, multiple substrains of BB rats exist, and lymphoid infiltrates are not limited to the pancreatic islets (201). The best-characterized BB colonies are those developed at the University of Massachusetts Medical School and now commercially distributed by Biomedical Research Models (Worcester, MA). A diabetes-resistant substrain (BBDR/Wor) has been selected, as have several diabetes-prone (BBDP/Wor) lines. These BB rats are fully inbred and characterized. In the Worcester virus-antibody-free (VAF) barrier colony, the frequency of type 1 diabetes in both sexes of DP rats exceeds 90%, whereas no spontaneous type 1 diabetes develops in VAF BBDR/Wor rats. A major genetic and phenotypic difference distinguishing the DR from the various DP strains is the presence in the DP line of a recessive mutation, lymphopenia (Lyp), on Chr 4 (202,203,204). Although DP rats develop a thymus of normal size and cellularity, recent thymic T lymphocyte emigrants to the periphery [phenotypically Thy-1+ and not yet expressing a cell-surface adenosine diphosphate (ADP)-ribosyltransferase (formerly designated RT6 and now designated ART2)] are very short-lived (205,206). The consequence is a profound T-lymphopenia/lymphocytopenia. Hence, these rats are severely immunocompromised, as reflected by impaired T-lymphocyte function in vitro, impaired ability to reject foreign tissue grafts, increased susceptibility to B-lymphomagenesis, and requirement for special husbandry procedures to protect against infections by opportunistic pathogens. This obviously contrasts sharply with NOD mice, whose T lymphocytes are long-lived in the periphery, present in large numbers, and express a functional ortholog of rat RT6 (now designated as ART2 in both genera) (207). Whereas BBDP rats are strongly immunocompromised, as evidenced by difficulty in rejecting non-islet allografted tissues and being susceptible to infectious pathogens, NOD T cells are capable of rapidly rejecting allografted tissues, and NOD mice are remarkably pathogen-resistant. There are many additional distinctions between BB rats and NOD mice in the diabetes syndrome itself, reviewed elsewhere, and some are summarized in Table 18.4 (6,197,198).
TABLE 18.4. Comparison of Differences Between Three Rodent Models of Type 1 Diabetes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
GENETICS OF TYPE 1 DIABETES
Superficially, the genetic basis for the spontaneous development of type 1 diabetes in rat models appears less complex than that in the NOD mouse or in humans. However, this may relate to the level of study of the process, since initially the genetics of type 1 diabetes in NOD mice was also inferred to be less complex, and possibly only oligogenic (100,102). As in humans and mice, the detection of “Idd” loci in the rat genome has been facilitated by the development of polymorphic microsatellite markers that can now be scored by automated methods (201). Segregation analysis between BBDP crossed with other type 1 diabetes-resistant strains have established that the MHC class II alleles encoded by the RT1u haplotype of the BB rat are essential for diabetogenesis (201,208,209,210,211). This MHC susceptibility, designated “Iddm2,” is permissive for insulitis in the heterozygous state; however, most diabetic segregants are MHC homozygotes. Analyses of insulitis and development of type 1 diabetes following outcross of diabetes-prone BB/Wor rats of the “NB” subline to RT1u congenic stocks of PVG rats carrying recombinants in the class I versus class II region have established the requirement for the RT1-Du/Bu alleles at the two class II loci (homologues of human DQ and DR, respectively) (210). Unlike
the nonexpression of H2-E molecules on APC of the NOD mouse, APC of the BB rat do express the DR homologue (RT1-Du). Sequence analysis of the BB rat DQ homologue (RT1-Bu) showed it differed from both the high diabetes risk-conferring human HLA-DQβ alleles and the NOD H2-Abg7 molecule. In contrast to NOD mice and humans, the BB rat allele contains an aspartic acid at residue 57 of the β chain instead of exhibiting “diabetogenic” non-Asp substitutions (212,213). All RT1u-expressing rat strains carry the requisite class II “Iddm2” alleles. Class I alleles from non-RT1u-positive rat strains apparently will substitute for the RT1-Au, Eu, and Cu class I alleles. Interestingly, although insulitis and thyroiditis were suppressed in class II heterozygous segregants in one outcross/backcross analysis, thyroiditis (another characteristic immune pathology of the BBDP-NB subline used in the outcross) was not (210).
the nonexpression of H2-E molecules on APC of the NOD mouse, APC of the BB rat do express the DR homologue (RT1-Du). Sequence analysis of the BB rat DQ homologue (RT1-Bu) showed it differed from both the high diabetes risk-conferring human HLA-DQβ alleles and the NOD H2-Abg7 molecule. In contrast to NOD mice and humans, the BB rat allele contains an aspartic acid at residue 57 of the β chain instead of exhibiting “diabetogenic” non-Asp substitutions (212,213). All RT1u-expressing rat strains carry the requisite class II “Iddm2” alleles. Class I alleles from non-RT1u-positive rat strains apparently will substitute for the RT1-Au, Eu, and Cu class I alleles. Interestingly, although insulitis and thyroiditis were suppressed in class II heterozygous segregants in one outcross/backcross analysis, thyroiditis (another characteristic immune pathology of the BBDP-NB subline used in the outcross) was not (210).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


