Melanie Jane Maslow, Cynthia Portal-Celhay
Rifamycins
The rifamycins were isolated by Sensi and colleagues at the Dow-Lepetit Research Laboratories in Milan in 1959 as a mixture of five substances from fermentation cultures of the organism now classified as Amycolatopsis mediterranei, which belongs to the Actinomycetaceae. Nicknamed “Rififi” after a French crime movie, the official name became rifamycin.1 Rifamycin SV, the first rifamycin used clinically in 1963, was replaced by rifampicin (rifampin) in 1968, owing to its improved bioavailability and greater activity against gram-positive and gram-negative bacteria and, especially, Mycobacterium tuberculosis. Rifampin is increasingly utilized as an adjunct for foreign-body infections associated with biofilm production, in combination with other antibiotics for infections due to multidrug resistant gram-negative bacilli, and for infections caused by intracellular pathogens.2 The newer rifamycins have fewer drug interactions and improved pharmacokinetics, resulting in shorter treatment courses for tuberculosis. Some of the newer rifamycins have an expanded spectrum of activity and unique pharmacokinetics and are used for the treatment of Clostridium difficile infection, hepatic encephalopathy, traveler’s diarrhea, and irritable bowel syndrome. Novel investigational rifamycins and new delivery systems are in development.
Structure and Mechanism of Action
The rifamycins belong to the ansamycin family (from the Latin ansa, meaning “handle”) of antimicrobial agents, so named because of their basket-like structure containing an aliphatic chain connecting two ends of a naphthoquinone core. Rifampin is the 3-(4-methyl-1-piperazinyl)-iminomethyl derivative of rifamycin SV. The structures of the four rifamycins currently approved in the United States—rifampin, rifabutin, rifapentine, and rifaximin—are shown in Figure 27-1.
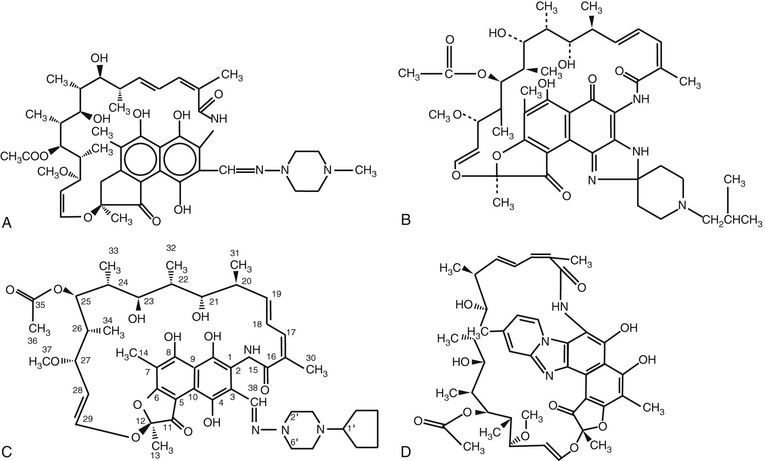
The antibacterial action of the rifamycin derivatives results from high-affinity binding to the DNA-dependent RNA polymerase of prokaryotes (RNAP) and inhibition of RNA synthesis. RNAP is a complex enzyme with an α2ββ′ω-subunit structure. The structure of the Thermus aquaticus core RNAP in complex with rifampin determined at 3.3-Å resolution showed that rifampin binds to a site on the β-subunit deep within the DNA/RNA channel, in a position that physically gets in the way of the growing oligonucleotide chain after the first- or second-chain elongation step.3 The structure, combined with biochemical results, led to a mechanistic model for inhibition of RNAP in which rifamycins sterically block synthesis of an RNA product longer than three nucleotides (“steric-occlusion model”). More recently, Artsimovich and associates4 proposed a new mechanism for inhibition of RNAP by rifamycins that operates in addition to, or instead of, the steric-occlusion mechanism. Their model proposes that rifamycins also inhibit transcription at earlier steps through an allosteric signal that disfavors binding of Mg2+ ion in the active site, thus slowing down catalysis and facilitating dissociation of short RNA transcripts. However, the allosteric model remains controversial, because others have shown that rifamycins do not affect the affinity of binding of Mg2+ to the RNAP active center.5
Mechanisms of Resistance
Rifampin possesses highly effective bactericidal action against M. tuberculosis and is used as a first-line regimen against this agent. However, pathogens develop resistance to rifampin at a rate of 10−8 to 10−9 per bacterium per cell division. Mutations conferring rifampin resistance are confined almost exclusively to the rpoB gene in most organisms and result in a decreased affinity of the RNAP to the antibiotic. More than 95% of M. tuberculosis clinical strains resistant to rifampin harbor a mutation in an 81-base pair region of the rpoB gene known as the rifampin resistance determining region (RRDR) that includes codons 507 to 533 and encodes 27 amino acids.6 Importantly, most of the strains have alterations at codon 526 that result in amino acid replacements or have missense mutations in codon 531. Studies with patient isolates have confirmed and extended these findings.7,8 Studies designed to determine the relationship between rpoB structural changes and rifampin minimal inhibitory concentrations (MICs) have found a strong correlation. In particular, amino acid substitutions at positions 526 and 531 conferred high-level resistance to rifampin, rifabutin, and rifapentine.9 The molecular mechanism in the 4% of the rifampin-resistant M. tuberculosis isolates that lack RRDR changes is unknown.
Alternative mechanisms of rifampin resistance also have been observed. Pseudomonas fluorescens has impaired cellular uptake of rifampin. Antibiotic modification occurs in strains of Mycobacterium smegmatis that carry the arr gene, causing ribosylation of rifampin and decreased susceptibility. An arr2 gene, similar to that in M. smegmatis, has been identified on a plasmid in clinical isolates of Klebsiella pneumoniae, Pseudomonas aeruginosa, and Enterobacteriaceae. Finally, the enzyme from some species, such as Nocardia, is intrinsically less susceptible to rifampin inhibition because of metabolic changes at the critical hydrogen-binding sites.10
Delineation of the molecular mechanisms of rifamycin resistance has resulted in the development and application of several polymerase chain reaction (PCR)-based strategies designed to rapidly detect mutations associated with rifampin resistance. One of the latest assays, Xpert MTB/RIF, a hemi-nested PCR, detects M. tuberculosis and rifampin resistance by amplifying five overlapping probes complementary to the RRDR of the rpoB gene and subsequently probes this region for mutations associated with rifampin resistance.11,12 Compared with conventional diagnosis methods, the Xpert MTB/RIF assay can detect M. tuberculosis and rifampin resistance in one sputum sample within 2 hours, is nearly fully automated, and requires minimal biosafety measures. The Xpert MTB/RIF assay has been validated by several studies as a highly sensitive and specific detection test for the diagnosis of tuberculosis and rifampin resistance.13 Other molecular assays to detect rifampin resistance are commercially available, such as the GenoType MTBDR assay 14 and the INNO-LiPA Rif assay.15 However, both tests have lower sensitivity on clinical specimens compared with culture isolates. More novel diagnostic tests, such as TB ID/R,16 are being configured in a low-cost test platform to provide rapid diagnosis and drug susceptibility information for tuberculosis in the developing world and in countries dealing with a dual burden of human immunodeficiency virus (HIV) infection and multidrug-resistant tuberculosis, where the need is acute.
Antimicrobial Activity
Although most commonly associated with the treatment of tuberculosis, the rifamycins exhibit broad antibacterial activity. Rifampin is most potent against gram-positive bacteria, including Staphylococcus aureus, coagulase-negative staphylococci, Streptococcus pyogenes, Streptococcus pneumoniae, Streptococcus viridans, C. difficile, and Listeria monocytogenes. Gram-negative bacteria against which rifampin is effective include Haemophilus influenzae, Neisseria meningitidis, and Helicobacter pylori.17,18 Rifampin has shown synergistic activity with other agents against multidrug resistant bacteria, especially P. aeruginosa and Acinetobacter baumannii. In addition to potent activity against M. tuberculosis, rifamycins have activity against nontuberculous mycobacteria, including Mycobacterium avium-intracellulare complex (MAC), Mycobacterium kansasii, and Mycobacterium marinum.19 Intracellular pathogens against which it is active include Chlamydia, Legionella, Brucella, and Bartonella species.20,21 The MICs of representative organisms to rifampin are shown in Table 27-1.
TABLE 27-1
Antimicrobial Activity of Rifampin
| SPECIES | MIC* RANGE (µg/mL) | MIC90 (µg/mL) | MIC50 (µg/mL) |
| Staphylococcus aureus | 0.008-0.015 | 0.015 | 0.015 |
| Staphylococcus epidermidis | 0.004-0.015 | 0.015 | 0.015 |
| Group A streptococcus | 0.03-0.12 | 0.12 | 0.12 |
| Group B streptococcus | 0.25-1 | 1 | 1 |
| Streptococcus pneumoniae | 0.06-32 | 0.12 | 0.12 |
| Streptococcus viridans | 0.03-8 | 0.06 | 0.12 |
| Enterococcus faecalis | 1-8 | 2 | 8 |
| Haemophilus influenzae | 0.5-64 | 1 | 1 |
| Neisseria meningitidis | 0.015-1 | 0.03 | 0.5 |
| Listeria monocytogenes | ≤0.12-0.25 | ≤0.12 | 0.25 |
| Escherichia coli | 8-16 | 8 | 16 |
| Klebsiella pneumoniae | 16-32 | 32 | 32 |
| Proteus mirabilis | 4-8 | 4 | 8 |
| Enterobacter cloacae | 16-64 | 64 | 64 |
| Acinetobacter spp. | 4-16 | 8 | 8 |
| Clostridium difficile | 0.10->100 | 1.56 | >100 |
| Pseudomonas aeruginosa | 32->64 | 32 | 64 |
| Mycobacterium tuberculosis | 0.06-0.25 | 0.25 | 0.25 |
| Mycobacterium avium-intracellulare | 0.78->100 | 6.25 | 50 |
| Mycobacterium kansasii | 0.025-3.13 | 0.2 | 3.13 |
| Mycobacterium marinum | 0.1-0.39 | 0.2 | 0.39 |
| Mycobacterium fortuitum | 12.5-100 | 50 | 100 |
| Mycobacterium scrofulaceum | 0.1-6.25 | 0.78 | 6.25 |
| Legionella spp. | 0.001-0.5 | ≤0.25† | 0.125 |
| Bartonella spp. | 0.03-0.06 | 0.12 | 0.25 |
| Chlamydia spp. | 0.0075-0.03 | 0.015 | 0.031 |
| Brucella spp. | 0.5-1 | 2 | 4 |
| Helicobacter pylori | 0.125-0.75 | 0.125 | 0.125 |
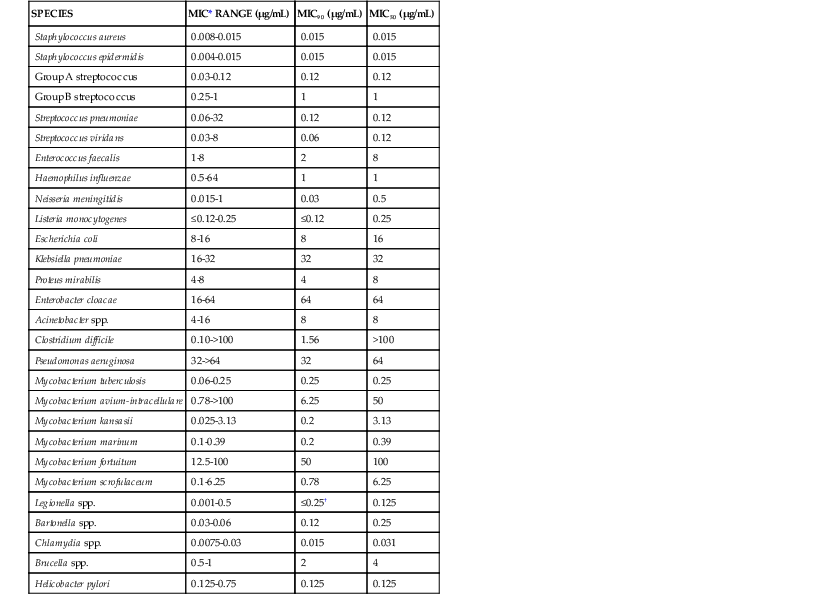
* MIC ≤ 2 µg/mL is considered susceptible.
† Geometric mean MIC.
MIC, minimal inhibitory concentration; MIC50, minimal inhibitory concentration for 50% of isolates; MIC90, minimal inhibitory concentration for 90% of isolates.
Pharmacokinetics
The rifamycins are highly lipophilic compounds that share a common chemical structure but differ in their pharmacokinetic properties,22 which are summarized in Table 27-2. Rifampin is dosed daily as 600 mg in adults and 10 to 20 mg/kg in children. The drug undergoes rapid and complete absorption, which is improved if taken on an empty stomach. Rifampin is the only rifamycin that has an intravenous formulation. It is widely distributed to most body tissues and fluids including the cerebrospinal fluid (CSF), where doses of 20 mg/kg in children achieve mean CSF levels of 2 µg/mL.23 Concentrations in bone are similar to or exceed serum concentrations,24 and the drug crosses the placenta and enters breast milk. Rifampin undergoes an enterohepatic circulation and deacetylation with rapid elimination in bile. Urinary excretion is 13% to 24% with no dose adjustment necessary in renal insufficiency. However, dose adjustment is recommended with severe hepatic insufficiency. The rifampin substrate is red, and the high lipophilic properties and wide distribution often turn body fluids such as urine, tears, sweat, feces, and contact lenses red-orange. Rifampin, rifabutin, and rifapentine have long postantibiotic effects against M. tuberculosis of 68 to 75 hours.25
TABLE 27-2
Comparative Pharmacokinetics of the Rifamycins in Healthy Adults
| DRUG | BIOAVAILABILITY (%) | tmax (hr) | Cmax (µg/mL) | t1/2 (hr) | PROTEIN BINDING (%) |
| Rifampin | 68 | 1.5-2 | 8-20 | 2-5 | 80 |
| Rifabutin | 20 | 2.5-4 | 0.2-0.9 | 32-67 | 72-85* |
| Rifapentine | 70 with food | 4.8-6.6 | 8-30 | 14-18 | 98 |
| Rifaximin | <0.4 | 0.8-1 | 3.4 ± 1.6 ng/mL | 1.8-4.8 | 62-67.5 |
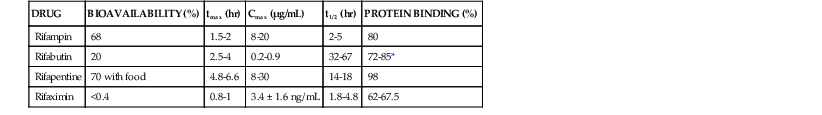
* From Blaschke TF, Skinner MH. The clinical pharmacokinetics of rifabutin. Clin Infect Dis. 1996;22(suppl 1):S15-S21.
Cmax, peak serum concentration; ng, nanograms; tmax, time to Cmax; t1/2, serum half-life.
Modified from Burman WJ, Gallicano K, Peloquin C. Comparative pharmacokinetics and pharmacodynamics of the rifamycin antibacterials. Clin Pharmacokinet. 2001;40:327-341.
Rifabutin is a spiropiperidyl rifamycin analogue with a relatively slower and larger volume of distribution compared with rifampin and a long terminal half-life, permitting once-daily dosing. Food does not affect the area under the concentration-time curve (AUC) but increases the peak concentration. Rifabutin is more lipid soluble than rifampin and extensively distributed throughout the body, with a CSF concentration one half that of serum after a 450-mg dose.26 The extensive redistribution of rifabutin in tissues leads to minimum plasma concentrations of 10% to 15% of the peak 24 hours after an oral dose. The two active metabolites are 25-O-desacetyl rifabutin and 31-OH-rifabutin. Fifty-three percent of the dose is excreted in urine as the primary active C-25 desacetyl metabolite, and dose reduction is recommended for a creatinine clearance less than 30 mL/min.27 Rifabutin should be administered with caution in patients with severe hepatic disease.
Rifapentine, a cyclopentyl rifamycin, is a more potent and longer-acting rifamycin.28 Food increases the absorption, and a high-fat meal results in a 50% increase in serum concentration. It is therefore recommended that rifapentine be taken with a meal. Rifapentine achieves high intracellular concentrations, exceeding that of rifampin. Concentrations in the CSF are undetectable.28 Less than 10% of rifapentine is excreted in urine unchanged.22 Rifapentine does not induce its own metabolism. The primary route of metabolism, mediated by an esterase enzyme, is nonoxidative to the 25-desacetyl metabolite.
Rifampin, rifabutin, and rifapentine are metabolized in hepatocytes and intestinal microsomes to deacetylated, hydroxylated, and formyl derivatives.22 The parent drug and the active 25-O-deacetyl metabolite are found in serum, excreted in bile, and eliminated in feces. Cytochrome P450 (CYP) 3A is the major isozyme involved in the hydroxylation of rifabutin.22 Repeated administration of rifampin and rifabutin, but not rifapentine, results in autoinduction by gut or hepatic metabolism and increasing biliary secretion. This results in a decrease in the AUC of both drugs and a decrease in the half-life of rifampin.
Rifampin has the greatest effect on induction of hepatic and intestinal enzymes, especially CYP3A, followed by rifapentine, with rifabutin having the weakest effect. Induction is responsible for the majority of rifamycin drug interactions. The maximal effect occurs approximately 1 week after initiation of therapy. The serum concentration of rifampin is rarely affected because the metabolism is not mediated by CYP, but rifabutin is metabolized by CYP3A and bidirectional interactions have been observed.
Rifaximin has an additional ring linking the C-3 and C-4 positions. Absorption after an oral dose is minimal secondary to low intestinal permeability and water solubility. Less than 0.01% of a rifaximin dose is detectable in plasma, and 97% is recovered unchanged in the stool. Fecal concentrations were 8000 µg/mL in one study.29 The AUC increases 10 to 20 times, and the Cmax increases 6 to 10 times in patients with cirrhosis.30 Rifaximin can induce cytochrome P450 3A4, but no significant drug interactions have been reported secondary to the extremely low systemic concentrations. Although caution is recommended in using rifaximin in patients with Child-Pugh class C cirrhosis, no dosage adjustments are required.
Drug Interactions
Most drug interactions with the rifamycins result from induction of metabolic enzymes, especially CYP3A4, in the liver and small intestine. Rifampin and the other rifamycins have numerous interactions, outlined in Table 27-3, that usually result in a decrease in the AUC or Cmax of the concomitant drug, requiring dose adjustments. Rifampin also induces CYP1A2, CYP2C, and CYP2D6 and some drug transporter proteins, including P-glycoprotein, a transmembrane protein that acts as a cellular efflux pump. The HIV protease inhibitors are especially problematic when used with rifampin, with significant decreases in AUC.
TABLE 27-3
Rifamycin Drug Interactions
Endocrine Agents Neuropsychiatric and Neurologic Agents Opiates/Analgesics |

* Posaconazole and voriconazole recently added.
† Efavirenz, nevirapine, etravirine, rilpivirine.
‡ Rifabutin preferred for use with HIV protease inhibitors in decreased doses; HCV protease inhibitors contraindicated with rifampin and rifabutin.
§ Includes bosentan, ambrisentan, and atrasentan.
‖ Reported with rifampin only.
HCV, hepatitis C virus; HIV, human immunodeficiency virus; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; NNRTIs, non-nucleoside reverse-transcriptase inhibitors.
Modified with permission from Baciewicz AM, Chrisman CR, Finch CK, et al. Update on rifampin, rifabutin, and rifapentine drug interactions. Curr Med Res Opin. 2013;29:1-12; and Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. Tables 14, 15a, 15b, and 15e. Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf. 2011.
The ability to induce metabolic enzymes, especially CYP3A4, is greatest with rifampin, intermediate with rifapentine, and least with rifabutin.22 When used with HIV protease inhibitors, this leads to an increase in the rifabutin level and decreased doses are required.22 Rifapentine also induces CYP3A4 and CYP2C and can interact with the same drugs as rifampin. The degree of interaction correlates with the dose and frequency of administration. Rifapentine is not recommended for use in combination with the HIV protease inhibitors and may be associated with decreased absorption in HIV-infected patients.22
The hepatitis C virus protease inhibitors boceprevir and telaprevir are contraindicated with the rifamycins because of an increase in the plasma concentrations of the rifamycin and a decrease in the plasma concentrations of the protease inhibitors. A recent comprehensive update on rifamycin drug interactions and recommendations for dosing was done by Baciewicz and colleagues31 and can also be found in the antiretroviral interactions section of the most recent U.S. Department of Health and Human Services guidelines.32
Rifabutin concentrations can be significantly increased, and there may be complex bidirectional interactions with concomitant administration of other medications, particularly in patients infected with HIV who are taking antiretroviral agents. Some of the major interactions are seen with fluconazole, ketoconazole, clarithromycin, and all of the HIV protease inhibitors.33
Adverse Drug Reactions
The major rifamycin adverse reactions can be divided by association with daily versus intermittent administration. The rifamycins have similar side effect profiles, except for rifabutin, which may cause uveitis and polyarthralgias. Despite the adverse reactions, the overall incidence of drug discontinuation was 1.9% in a large retrospective study, with discontinuation in over half the cases judged to be unnecessary.34
Allergic reactions to rifampin are infrequent, ranging from 0.01% to 0.26%, and often occur in patients previously treated or with intermittent dosing. Type I hypersensitivity reactions mediated by IgE antibodies can occur with symptoms ranging from angioedema, bronchospasm, and urticaria to shock. Anaphylaxis with symptoms can occur within minutes of the dose or be associated with prodromes, such as rash, from prior or intermittent dosing. Patients with HIV infection had poorer outcomes, and anti-rifampin serum IgE antibodies were detected.35
The rifampin-associated flulike syndrome consists of fever, chills, malaise, myalgias, and headache several hours after dosing and usually resolves within several hours. Daily rifampin dosing at 600 mg is well tolerated, although HIV-infected patients experience a higher incidence of side effects with higher dosages that may be due to a direct toxic effect of the drug in this population.35 The incidence of flulike syndrome escalates with higher doses (900 to 1200 mg) given weekly and can be as high as 54%.35 The flulike syndrome usually develops after 3 months of therapy and is rarely associated with thrombocytopenia, hemolysis, and renal failure. The etiology is believed due to circulating immunoglobulin (Ig) G (IgG) or IgM immune complexes that fix complement on endothelial cells or bind on the surface of cytokine-producing cells.35 After a change to weekly dosing or intermittent therapy, a more robust immune response may be seen on rechallenge. Switching to daily rifampin therapy usually results in disappearance of all symptoms. Thrombocytopenia and hemolysis are also likely immune mediated with formation of antigen–antibody complexes that bind to erythrocytes and platelets. Recent reports highlight newly observed adverse reactions, including recurrent disseminated intravascular coagulation, with intermittent rifampin dosing.36
Acute renal failure from rifampin usually occurs with intermittent dosing and presents as acute interstitial nephritis with tubular necrosis. Less common presentations are rapidly progressive glomerulonephritis and light chain proteinuria with polyclonal κ and λ chains. Most cases are secondary to adhesion of anti-rifampin antibodies to the renal tubular epithelium with subsequent complement binding. Recovery is usually complete after stopping the drug.37 Hemolysis and thrombocytopenia may occur in conjunction with renal toxicity.
Rifampin can cause changes in liver function, but serious injury is usually confined to patients with underlying liver disease due to alcohol, viral hepatitis, and other hepatotoxins, such as isoniazid, which often is co-prescribed. Long-term therapy with rifampin is associated with minor, transient elevations in serum aminotransferase levels in 10% of patients. Liver injury from rifampin is primarily cholestatic. In most patients, serum bilirubin levels increase during the first few days of therapy and normalize spontaneously. Within a few weeks of starting rifampin in patients with underlying liver disease, there may be a significant increase in direct and total bilirubin without evidence of liver injury. The bilirubin elevation is attributed to inhibition of bilirubin excretion and may be related to genetic defects in MRP2, the major bilirubin glucuronide transporter in hepatocytes. There are many reports of hepatitis and jaundice in patients with tuberculosis treated with rifampin, isoniazid, and pyrazinamide. However, the actual incidence of hepatotoxicity from rifampin is 1.1%.38 When rifampin is combined with isoniazid there is an increased incidence of hepatotoxicity. One explanation is an additive hepatotoxic effect of these drugs; however, a synergistic effect is favored. The induction of isoniazid hydrolase by rifampin is believed to lead to increased production of hydrazine, which can be activated by rifampin-induced CYP to hepatotoxins. This is seen more frequently in slow acetylators with the N-acetyltransferase 2 genotype.39 It is recommended that all antituberculous medications be discontinued if hepatotoxicity occurs. If the patient is severely ill, an option is to start three new medications (often a fluoroquinolone, streptomycin, and ethambutol) until liver function normalizes, at which time the original drugs are introduced one at a time, usually starting with rifampin.40
Severe hepatitis has been reported in 1.2% to 13% of patients treated with rifampin and pyrazinamide for latent tuberculosis infection, leading to updated recommendations regarding use and monitoring of this combination.41 A single nucleotide polymorphism in the gene encoding the organic anion transporting polypeptide 1B1 that plays a role in transporting bile acids and rifampin has recently been identified with the *15 haplotype, a predisposing factor to rifampin-induced liver injury.42
Gastrointestinal symptoms from the rifamycins are nausea, vomiting, diarrhea, and abdominal pain. Headache, fever, and pruritus can be seen. Rash may occur early in the course and resolve without discontinuation of the drug. More severe reactions include urticarial and diffuse rash, which may be accompanied by systemic symptoms. A lupus-like syndrome with detection of antinuclear antibodies has been reported with rifampin and rifabutin, often attributed to CYP drug interactions, and usually resolves after discontinuation of the drug.43
Rifapentine is better tolerated than rifampin, likely owing to higher protein binding. Once-weekly dosing is associated with a lower incidence of the flulike syndrome28; however, cases with hemolysis and renal failure have been reported. The incidence of hepatotoxicity is similar to rifampin. Hyperuricemia has been reported with rifapentine in up to 21.3% of patients.28
Rifabutin has a unique toxicity profile, including uveitis, leukopenia, and polyarthralgias. In a study of elderly patients with pulmonary MAC treated with 600 mg of rifabutin with ethambutol, streptomycin, and a macrolide, adverse events were seen in 77%, requiring dose adjustment in over half.44 The most common event was leukopenia, followed by gastrointestinal symptoms, abnormal liver enzymes, a diffuse polyarthralgia syndrome in 19%, and anterior uveitis in 8%. Uveitis was seen in patients receiving clarithromycin and is likely secondary to a drug interaction that raises rifabutin serum concentrations. Uveitis may mimic an infectious endophthalmitis with the presence of a hypopyon and has been reported in both HIV-infected and non–HIV-infected patients with pulmonary MAC infection with the 600-mg dose. Cases typically occur after several months of therapy, predominantly involve the anterior chamber, and respond to corticosteroids and cycloplegics. The etiology is believed to be an immunologic reaction to cell wall proteins of dead organisms, although a direct toxic effect of rifabutin has been suggested.45 Uveitis has been seen in HIV-infected patients receiving 300-mg dosing for prophylaxis as late as 14 months after initiation,46 often in combination with fluconazole or azithromycin or both. The flulike syndrome has been reported with intermittent therapy. Hepatitis, like that with rifampin, is more likely from concomitant use of hepatotoxins.
Rifaximin is associated with few adverse effects. An unusual case of rifaximin-associated neutropenia was described in a patient receiving 1200 mg daily with severe ulcerative colitis and a transjugular intrahepatic portosystemic shunt, suggesting increased absorption and decreased metabolism.47 The investigational drug rifalazil can cause arthralgias, fever, myalgias, taste perversion, transient neutropenia, and flulike symptoms.48
Immune-Modulating Effects of Rifampin
In addition to its bactericidal activity, rifampin has immune-modulating properties. An in vitro study showed inhibition of interleukin (IL)-1β and tumor necrosis factor-α (TNF-α), with increases in IL-6 and IL-10 when rifampin was added to stimulated human monocytes.49 Incubation of rifampin with proinflammatory cytokines in alveolar cells and human epithelial liver cells (HepG2) led to increased production of nitric oxide and increases in IL-8, interferon-γ (IFN-γ), and IL-β1.50 Cytoprotective effects of rifampin have also been observed. Pretreatment of cells derived from a human lung carcinoma with rifampin before infection with A. baumannii significantly decreased the rate of cell death by reducing oxidative stress and release of TNF-α and IL-6; this effect occurred in the absence of bactericidal activity.51 Pretreatment with rifampin in animal models of S. pneumoniae meningitis decreased neuronal loss by decreasing inflammation and free radical production in the CSF.52,53 Rifampin also decreased TNF-α production and inhibited macrophage apoptosis in M. tuberculosis infection.54 In vivo, patients with tuberculosis who were able to mount a higher IFN-γ response after 2 months of therapy had higher AUC0-8 values to rifampin, which predicted a disease-free outcome. It is unclear whether the higher rifampin concentrations led to a stronger IFN-γ response, but the authors suggest that IFN-γ may be a useful surrogate marker for predicting outcome in tuberculosis.55
Rifamycins for the Treatment of Tuberculosis
The treatment of active tuberculosis requires combination chemotherapy to avoid the selection of naturally occurring drug-resistant mutants. Since the introduction of rifampin 4 decades ago, the rifamycins have been cornerstone agents in the treatment of tuberculosis, particularly when it was shown that rifampin in combination therapy reduced the overall treatment time from 18 to 9 months.56 In addition, it has been shown that infections caused by rifampin-resistant strains of M. tuberculosis require a longer duration of treatment and are associated with higher rates of treatment failure than those caused by drug-susceptible strains.56 Rifampin is bactericidal against M. tuberculosis and among the first-line agents against tuberculosis has the most potent sterilizing activity, which is defined as the ability of a drug to kill persistent and/or metabolically dormant organisms. This may be in part due to its rapid onset of action.57 International guidelines recommend a maximum daily dose of 600 mg (10 mg/kg) for daily and intermittent treatment. However, these recommendations were based on historical pharmacokinetic,58,59 toxicity,60,61 and cost arguments62 and have largely been refuted by more recent scientific data.63 Data from in vitro systems and animal models provide compelling evidence that rifampin’s activity against M. tuberculosis is concentration dependent and correlates best with the quotient of the AUC and the MIC.63 Using the mouse model, Jayaram and co-workers found that the 10 mg/kg dose of rifampin used in humans falls at the low end of a steep and tall dose-response curve, indicating that increasing doses may produce log linear increases in bactericidal activity.64 These data are supported by small-scale studies that showed an early bactericidal activity using high doses (20 mg/kg) of rifampin.65,66 However, studies of early bactericidal activity reflect bactericidal and not necessarily sterilizing activity. Therefore, more studies are needed to evaluate the potential of a higher dose of rifampin to further reduce treatment duration to less than 6 months. Evidence of this potential is provided by the study of Kreis and associates, who evaluated a 3-month regimen with daily rifampin (1200 mg), isoniazid (900 mg), and streptomycin (1 g) and achieved near-complete sputum culture negativity after 90 days, without additional toxicity.67 Unfortunately, this study had several limitations. First, without pyrazinamide, the recurrence rate was 11.4% during the first year after treatment. Second, there was no direct comparison of daily administration of the current standard dose (600 mg) against daily administration of higher doses.
The primary toxicities attributed to rifampin are discussed in detail in the section on adverse reactions and are usually associated with intermittent dosing and not expected to occur more frequently with increased daily dosing. Transaminitis and increased bilirubin levels after starting treatment do not appear dose related,68 and hepatoxicity usually occurs with preexisting liver disease or with use of concomitant hepatotoxins.69 Systemic side effects such as the “flulike syndrome” are related to intermittent treatment rather than to the dose of rifampin.70 Furthermore, rifampin has been used at higher doses for nonmycobacterial infections such as brucellosis,71 staphylococcal infections,72 and cutaneous leishmaniasis,73 without reports of increased adverse effects, supporting the idea that the majority of rifampin’s adverse effects are idiosyncratic and not dose related.
A recent study has suggested that high-dose rifampin may play a role in infections caused by low-level resistant strains74 and may help prevent the emergence of drug resistance.75 Recent data also suggest that regimens containing higher doses of rifampin are safe and could be associated with a survival benefit in patients with severe disease.76
In summary, higher doses of rifampin may help reduce the duration of treatment of tuberculosis without a significant increase in adverse events. However, more extensive, dose-ranging, tolerability, and extended bactericidal activity studies of high doses of rifampin are warranted.
Rifapentine, with its more potent in vitro activity (MIC, 0.06 vs. 0.25 µg/mL)77 and longer serum half-life (11 to 18 hours vs. 2 to 4 hours) than rifampin,22,78 was developed with the goal to provide an option for once-weekly therapy for tuberculosis.79 The approved U.S. Food and Drug Administration (FDA) dose for the treatment of active tuberculosis is 600 mg administered once or twice weekly in combination with other antimycobacterial drugs. However, it has seen little clinical use because once-weekly rifapentine-isoniazid during the continuation phase of treatment was associated with greater drug-susceptible relapse among HIV-negative patients with lung cavitation80 and a significant incidence of acquired rifamycin monoresistance among HIV-positive patients.81
Recent work using the mouse model of antituberculosis chemotherapy has shown that regimens containing rifapentine administered 5 days per week can achieve durable cure without relapse after 3 months. Their results also indicated that rifapentine is four times more potent than rifampin in the murine model.82 These encouraging results prompted a randomized, multicenter, phase II clinical trial conducted by the Tuberculosis Trials Consortium (TBTC study 29) to determine the safety, tolerability, and antimicrobial activity of a regimen in which rifampin 10 mg/kg was replaced by rifapentine 10 mg/kg administered 5 days per week during the first 8 weeks (intensive phase) of combination treatment of pulmonary tuberculosis.83 Their results showed that the rifapentine regimen was safe but not significantly more active than a standard rifampin regimen by the surrogate end point of culture status at completion of the intensive phase. However, one limitation of this study was administration without food, which has been shown to increase the Cmax of rifampin84 but decreases rifapentine bioavailability.85
The optimal rifapentine dose and schedule for treating tuberculosis in humans have not been established, but the mouse model suggests that the bactericidal and sterilizing activity of rifapentine increases virtually without plateau up to doses of 160 mg/kg and that daily dosing improves outcomes. Dooley and colleagues recently conducted a rifapentine dose-escalation study in healthy adults and showed that daily rifapentine doses of up to 20 mg/kg administered with food were safe and produced rifapentine exposures exceeding those in mice receiving 10 mg/kg.86 Their results provided safety and tolerability data to support the need of future clinical trials of rifapentine at high daily doses for treatment of tuberculosis.
Rifabutin has a much longer half-life than rifampin (35 hours vs. 2 to 4 hours). In clinical trials, the efficacy of rifabutin administered daily or intermittently has been shown to be similar to that of rifampin.87,88 Rifabutin’s primary advantage in the treatment of tuberculosis is its reduced induction of hepatic metabolism (cytochrome P450 system) and its usefulness in patients with tuberculosis and HIV coinfection who are also receiving antiretroviral therapy. Rifampin markedly decreases serum concentrations of the HIV type 1 (HIV-1) protease inhibitors89 and has substantial effects on concentrations of non-nucleoside reverse-transcriptase inhibitors (NNRTIs).90 Therefore, rifabutin-based therapy was recommended by the Centers for Disease Control and Prevention as a way of avoiding serious drug-drug interactions and allowing use of rifamycins during treatment of tuberculosis in HIV-coinfected patients.91 However, Benator and colleagues raised the question of whether the use of rifabutin was a cause of acquired rifampin resistance in HIV-infected patients who had low CD4+ T-lymphocyte counts.80 Additional studies suggest that low concentrations of the rifamycin component related to intermittent dosing, rather than the specific agent, determine the risk for acquired rifamycin resistance.92
The most recent guidelines for the treatment of active tuberculosis in HIV-coinfected patients recommend that, despite pharmacokinetic drug interactions, a rifamycin should be included in regimens for patients receiving antiretroviral agents, with dosage adjustment if necessary. If a protease inhibitor–based regimen is used, rifabutin is the preferred rifamycin, but the dose should be decreased from 300 mg daily to 150 mg every other day (daily if given with atazanavir). If given with efavirenz, the rifabutin dose should be increased from 300 mg to 450 mg daily and the dose of efavirenz should remain unchanged.32
Recent data examined the tolerability of rifabutin when co-administered with other medications affected by rifampin’s induction of cytochrome P450, such as methadone and coumadin, and in patients who previously had a rifampin-related adverse effect. Their results showed that rifabutin is well tolerated and have limited the need for dosing adjustments of other medications. Their results also suggest that rifabutin could be considered as an alternative agent in patients who develop rifampin-related liver injury but that rifabutin should be used with caution in patients with other types of underlying liver disease.93 Data from tuberculosis-infected transplant patients receiving tacrolimus or cyclosporine also suggest that rifabutin should be used in this population instead of rifampin to avoid pharmacologic interactions and achieve a lower risk for acute graft rejection.94
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


