Retinoblastoma Syndrome
Timothy A. Damron
Retinoblastoma syndrome encompasses familial retinoblastoma and a number of secondary malignancies that may develop as a result of a “second hit” at the site of the RB1 gene, where a somatic mutation added to the germline RB1 mutation inactivates the tumor suppressor gene function. The synonym “retinoblastoma/osteogenic sarcoma syndrome” reflects the fact that osteosarcoma is the most common secondary tumor in these patients. Retinoblastoma is a malignancy of the embryonic neural retina.
Pathogenesis
Etiology
Prototypical example of “two-hit” theory for genetic predisposition to cancer
First hit: germline (familial form) or somatic (nonfamilial form) mutation of RB1 gene at 13q14.1
Second hit: somatic mutation in all cases at RB1 locus
Radiation increases the risk in a dose-dependent fashion above 5 Gy.
Resultant inactivation of RB1 tumor suppressor gene function is associated with retinoblastoma, post-retinoblastoma osteosarcomas, and other sarcomas (Box 7.4-1), and in some sarcomas not associated with retinoblastoma syndrome (breast and non–small-cell lung carcinoma).
Inheritance: autosomal dominant (AD) with almost full penetrance in familial form
Epidemiology
Retinoblastoma
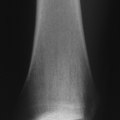
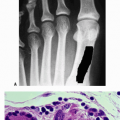
Frequency: 1/3,500 to 1/25,000 (Fig. 7.4-1)
Male:female equal
Secondary sarcoma
Relative risk compared to normal population: 30
Cumulative incidence over 50 years
Hereditary retinoblastoma: 51%
Nonhereditary retinoblastoma: 5%
Box 7.4-1 Secondary Tumors Associated with Retinoblastoma Syndrome
OsteosarcomaRelated posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree