Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .


Introduction
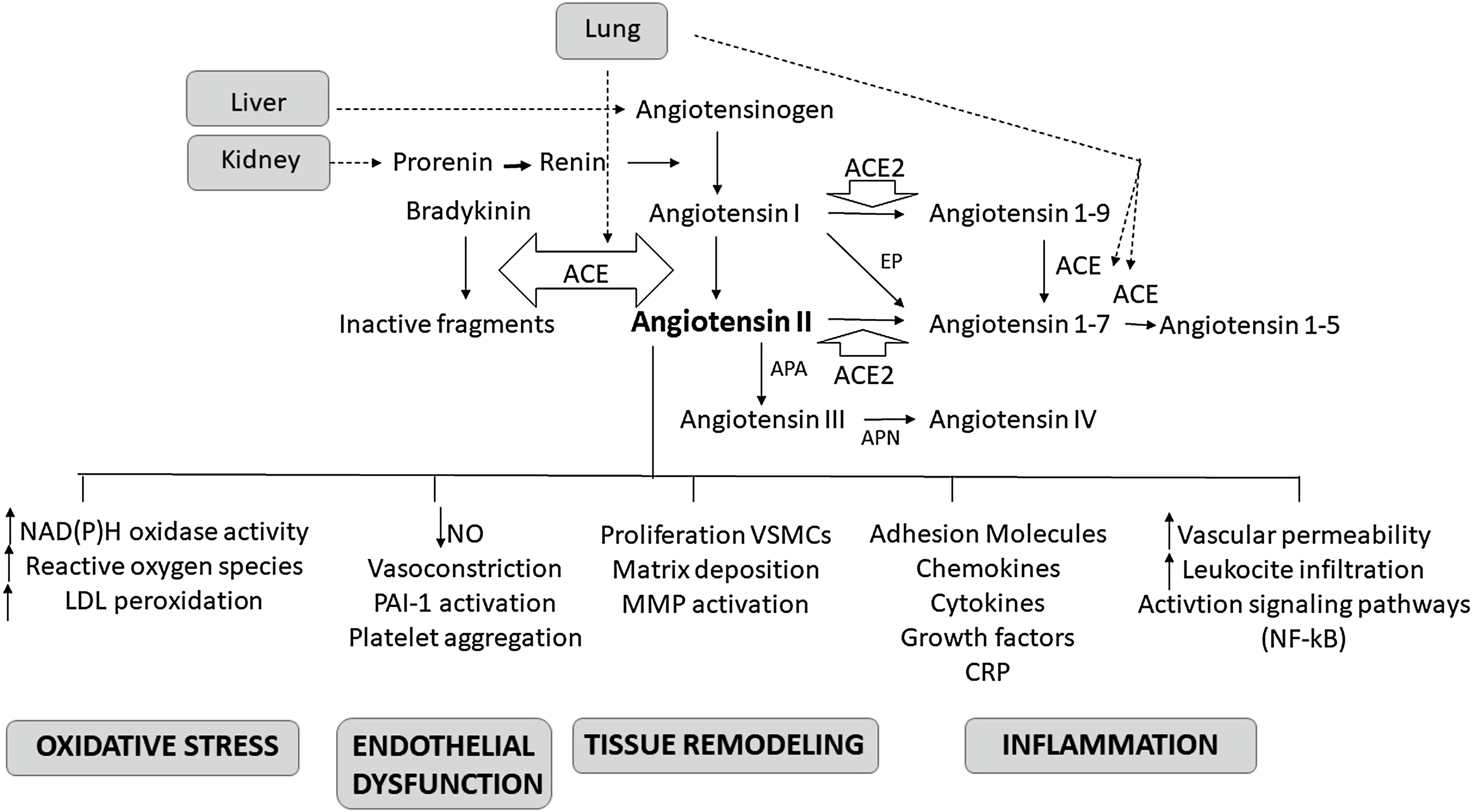
Arterial hypertension is a highly prevalent condition characterized by increasing incidence rate in the global population (which is expected to rise to 30% by 2025) [ ] and associated with important end-organ complications (heart diseases, cerebrovascular diseases, and renal diseases) [ , ]. The renin–angiotensin–aldosterone system (RAAS) plays a pivotal role in the pathogenesis of hypertension by regulating the systemic vascular resistance and circulatory volume through its effects on water and electrolyte balance. The renin agniotensin system is also involved in vascular inflammation and remodeling [ , ] ( Fig. 5.1 ). The adverse effects of chronic exposure to high concentrations of angiotensin II (Ang II) and aldosterone are outlined in Table 5.1 [ ]. Suppression of RAAS represents a key strategy in the treatment of hypertension, cardiovascular disease, and chronic renal disease. The aim of this chapter is to elaborate the physiologic role of RAAS and to evaluate the effects of RAAS modifying therapeutic agents.
| Adverse effect | Direct effect of Ang II | Direct effect of Aldo |
|---|---|---|
| Myocardial remodeling (fibrosis, hypertrophy) | Yes | Yes |
| Vascular remodeling (fibrosis, hypertrophy) | Yes | Yes |
| Increase ROS | Yes | Yes |
| Proinflammatory | Yes | Yes |
| Arrhythmogenic | Yes | Yes |
| Vascular endothelial dysfunction | Yes | Yes |
| Systemic hypertension | Yes | Yes |
| Glomerular damage | Yes | Yes |
| Glomerular dysfunction (proteinuria) | Yes | Yes |
| Increased intraglomerular pressure | Vasoconstriction | Fluid retention and increase SNS activity |
| Tubulo-interstitial injury | Yes | Yes |
| Baroreceptor dysfunction | Yes | Yes |
| Sympathetic activation | Yes | Yes |
| Inotropy | Yes | No |
| Direct heart rate increase | Yes | No |
| Salt appetite | Yes | Yes |
| Increased thirst | Yes | No |
| Sodium and water retention | Yes | Yes |
| Potassium wasting | No | Yes |
Physiological aspects of RAAS
Renin is synthetized as prorenin in the juxtaglomerular epithelioid cells and either released as prorenin or further processed to form active renin, which is stored as granules. These granules are then released in a controlled manner. Conversely, angiotensinogen (a renin substrate) is constitutively released from the liver and is usually present in excess. Increased renin synthesis and release occur during low systemic blood pressure, hypovolemia, sodium deprivation, and sympathetic stimulation. In the circulation, renin metabolizes angiotensinogen liberating angiotensin I (Ang I). Angiotensin-converting enzyme (ACE), which is released from endothelial cells (mainly of the pulmonary vasculature), converts Ang I to Ang II. Alternative pathways involving aminopeptidases and non-ACE enzymes can also take part in the transformation of Ang I
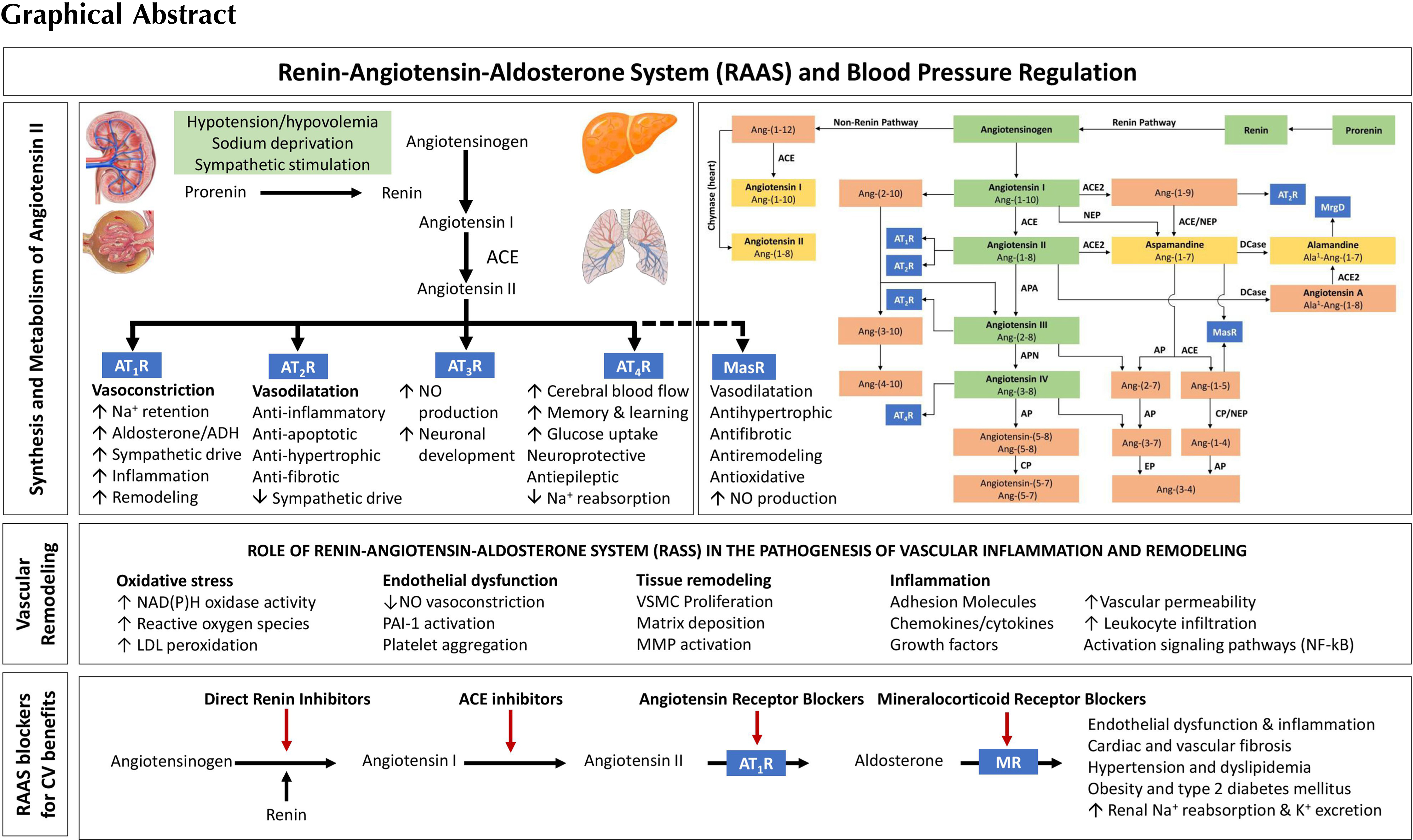
Four Ang II receptors (AT1R, AT2R, AT3R, and AT4R) have been identified to date [ ]. The action at the AT1R leads to increased sodium retention, vasoconstriction—especially that of the efferent arteriole of the kidney, stimulation of the thirst mechanism and desire for salt, enhanced sympathetic activation, and enhanced aldosterone release from the adrenal gland. The interaction of Ang II and this receptor mediates much of the changes associated with chronic RAAS activation summarized in Table 5.1 including ventricular hypertrophy, inflammation, fibrosis, structural remodeling, and cardiac dysfunction. To counterbalance the adverse effects of chronic RAAS activation, natriuretic peptides (NPs) are released to facilitate salt and water excretion and to cause vasodilatation [ ]. Neprilysin is a degrading enzyme for many of the NPs. Neprilysin inhibition alone is not sufficient to counterbalance the chronic RAAS activation that occurs in cardiovascular diseases like hypertension and heart failure. Hence, a combination of angiotensin receptor blocker and neprilysin inhibitor is used. Actions of AT2R stimulation are counterregulatory to those of the AT1R, leading to antiinflammatory, antifibrotic, and vasodilatory effects. The activation of AT3R leads to the production of nitric oxide (NO) and is responsible for neuronal development. The AT4R plays a role in regulating blood flow, inhibiting the reabsorption of sodium, in memory processes, and in vasodilatation.
Angiotensin I, II, III, and IV are also represented as Ang-(1–10), Ang-(1–8), Ang-(2–8), and Ang-(3–8), respectively [ ]. The discovery of a new angiotensin-converting enzyme, ACE2, that has 42% homology to ACE and is expressed in the heart, kidney, testis, endothelium of coronary arteries, and intrarenal vessels, and renal tubular epithelium introduced more complexity to our understanding about the RAAS [ ]. ACE2 acts as a counterregulatory molecule of the classical ACE effects, regulates the levels of Ang II, and limits its effects. The preferential substrate of ACE2 is Ang II, meaning that Ang II binds to ACE2 with a higher affinity than Ang I, leading to the formation of angiotensin 1-7 [Ang-(1–7)], also known as aspamandine [ ]. The latter molecule has pleiotropic effects that are mediated by a specific receptor (MasR) known as Mas-1. By binding to this Mas-1 receptor, the Ang-(1–7) induces vasodilation, natriuresis, and diuresis [ , ], the effects that oppose that of Ang 1 through AT1R. The binding of Ang I to ACE2 results in the formation of angiotensin 1–9 [Ang-(1–9)], which in turn can be converted to Ang-(1–7), by the action of ACE [ ]. The Ang-(1–7) or aspamandine can in turn be decarboxylated to form alamandine [ ]. The latter binds to Mas-related G-protein-coupled receptor member D (MrgD). The AT2R, MasR, and MrgD form the protective arm of the RAAS as they protect the kidney and heart through their antihypertensive actions [ ].
The terminal hormone of RAAS, aldosterone, exerts 90% of the mineralocorticoid activity of adrenal hormones and is a key regulator of sodium, potassium, and body fluid balance [ , , ]. Ang II level and an increased extracellular K + concentration increase the expression of the CYP11B2 gene which encodes aldosterone synthase [ ]. Acting via the mineralocorticoid receptor (MR), aldosterone modulates the expression of ion channels [epithelial sodium channel (ENaC)], pumps, and exchangers in the epithelial tissues. This leads to an increase in the transepithelial Na + and water reabsorption, and K + excretion. MRs are present not only in the kidneys, colon, salivary, and sweats glands, but also in the nonepithelial tissues such as the retina, brain, myocardium, vascular smooth muscle cells, macrophages, fibroblasts, and adipocytes [ ]. Since blood pressure is finely tuned by the RAAS, any imbalance in this system will produce arterial blood pressure alterations.
RAAS, inflammation, and remodeling
RAAS plays important roles in the initiation and maintenance of vascular inflammation and remodeling. Vascular inflammation leads to endothelial dysfunction. This facilitates migration of inflammatory cells into the vascular wall and stimulates smooth muscle cell proliferation and tissue injury, favoring atherosclerotic process. There is increasing evidence indicating a link between hypertension and atherosclerosis via Ang II-mediated inflammation. Animal and human studies show that Ang II has proinflammatory responses in the arteries, heart, and kidneys by regulating the expression of cytokines and chemokines. In human vascular smooth muscle cells, Ang II induces nuclear factor (NF)-κB (a proinflammatory factor downstream of tumor necrosis factor alpha—TNFα) activation and the expression of interleukin-6 (IL-6) [ ], while in vivo, it causes increased expression of Vascular Cell Adhesion Molecule-1 that is inhibited by administration of the AT1R antagonist losartan [ ]. Ang II plays a significant role in the initiation and progression of atherogenesis, an inflammation-mediated process. In injured arteries, Ang II favors recruitment of inflammatory cells inducing a feedback loop that produces more Ang II. It also induces the production of superoxide anions and activates the prooxidant NADH/ NADPH signaling [ ]. Ang II-mediated oxidative stress reduces the NO levels and activates the redox sensitive genes, particularly cytokines, adhesion molecules, and matrix metalloproteinases [ ]. Ang II is also a profibrotic factor. In animal studies, chronic infusion of Ang II increases the blood pressure and favors the inflammatory cell infiltration into the myocardium and induces cardiac fibrosis, oxidative stress, and endothelial dysfunction [ ]. The use of valsartan and fluvastatin, in atherosclerosis mouse model, can reduce the level of atherosclerotic lesions, superoxide anion, and the expression of Monocyte Chemoattractant Protein-1 and Intercellular Adhesion Molecule-1, indicating that blocking the inflammation and oxidative stress has beneficial effects on endothelium [ ]. Clinical studies show a reduction in cardiovascular events beyond blood pressure lowering. Thus, positively altering the endothelium and vascular wall structures, in turn, mediates a reduction in cardiovascular disease burden. The increase in NO bioavailability, mediated by several RAAS inhibitors, induces vasodilation; inhibits platelet aggregation, leukocyte adhesion to endothelium, DNA synthesis, and vascular smooth muscle cell proliferation, thus counteracting the oxidative stress and hypertension [ , ].
The proinflammatory and profibrotic effects of the RAAS are also mediated by aldosterone [ ]. In the vascular smooth muscle cells, aldosterone alters the insulin-like growth factor-1 receptor (IGF-1R) and modulates membrane structure via tyrosine kinase receptors. In the vascular endothelium, it decreases the synthesis of NO, and the formation of bone marrow-derived endothelial progenitor cells via oxidative stress and decreased levels of Vascular Endothelial Growth Factor Receptor 2 expression [ ]. In endothelial cells, aldosterone increases the expression of sodium channels (like ENaC), known as endothelial sodium channels (EnNaCs). The combination of raised aldosterone levels associated with high salt (sodium) intake leads to raised plasma sodium levels, stiffening of the cell cortex of the endothelial cells, decrease in endothelial (NO synthase-mediated) NO release, stiff endothelial cell syndrome (SECS), and impaired vascular function [ , ]. Thus, high aldosterone in combination with high salt intake may result in endothelial dysfunction which may contribute to a rise in blood pressure independent of the renal effects of aldosterone. Several molecular pathways activated by the interaction of aldosterone with the VSMC-MR and contributing to vascular remodeling have been described [ , ]. Effects of aldosterone may be also mediated by G-protein-coupled estrogen receptor-1 [ ]. The complexity of this system is due to the fact that other than systemic and local tissue RAAS, intracellular RAAS has been identified [ ], and that several other molecules such as ERK1/2, NADPH, and NF-κB may participate to activate the system or be activated.
RAAS blocking agents and cardiovascular protection in hypertension
The European Society of Cardiology/European Society of Hypertension (ESC/ESH) 2018 guidelines [ ] underline that blockade of RAAS should be part of the first therapeutic approach in combination with other antihypertensive drug classes for the management of patients with hypertension. The most widely used classes of drugs for this scope are as follows: (1) drugs inhibiting angiotensin-converting enzyme (ACEI), (2) antagonists of the AT1 receptors (ARBs), and (3) direct inhibitors of renin. Indirectly, we can also obtain the inhibition of renin release through β-blockers and α2-agonists. Last but not the least, the use of MR receptor antagonists blocking the actions of aldosterone on target tissues is also found to offer significant benefits in blood pressure control and cardiovascular protection.
ACE inhibitors
ACEIs are currently the most widely used agents for the treatment of hypertension; other cardiovascular diseases including heart failure and ischemic heart disease; and renal disease. These drugs act by inhibiting the ACE, thereby blocking the conversion of Ang I into Ang II. The antihypertensive activity of ACEI is the result of various effects: (a) the systemic and local tissue level inhibition of the effects of Ang II, with subsequent vasodilatation and reduction in plasma aldosterone levels, associated with increased natriuresis, diuresis, and reduction in left ventricular hypertrophy [ , ]; (b) increase in the circulating levels of bradykinin (with subsequent vasodilatation), prostacyclin and endothelium-derived hyperpolarizing factor, all of which induce vasodilation and inhibit vascular smooth muscle cells proliferation, and platelet adhesion [ , ]; (c) inhibitory effect on the release of antidiuretic hormone and reduction of both central and peripheral sympathetic activity [ ]; and (d) antioxidant activity [ ].
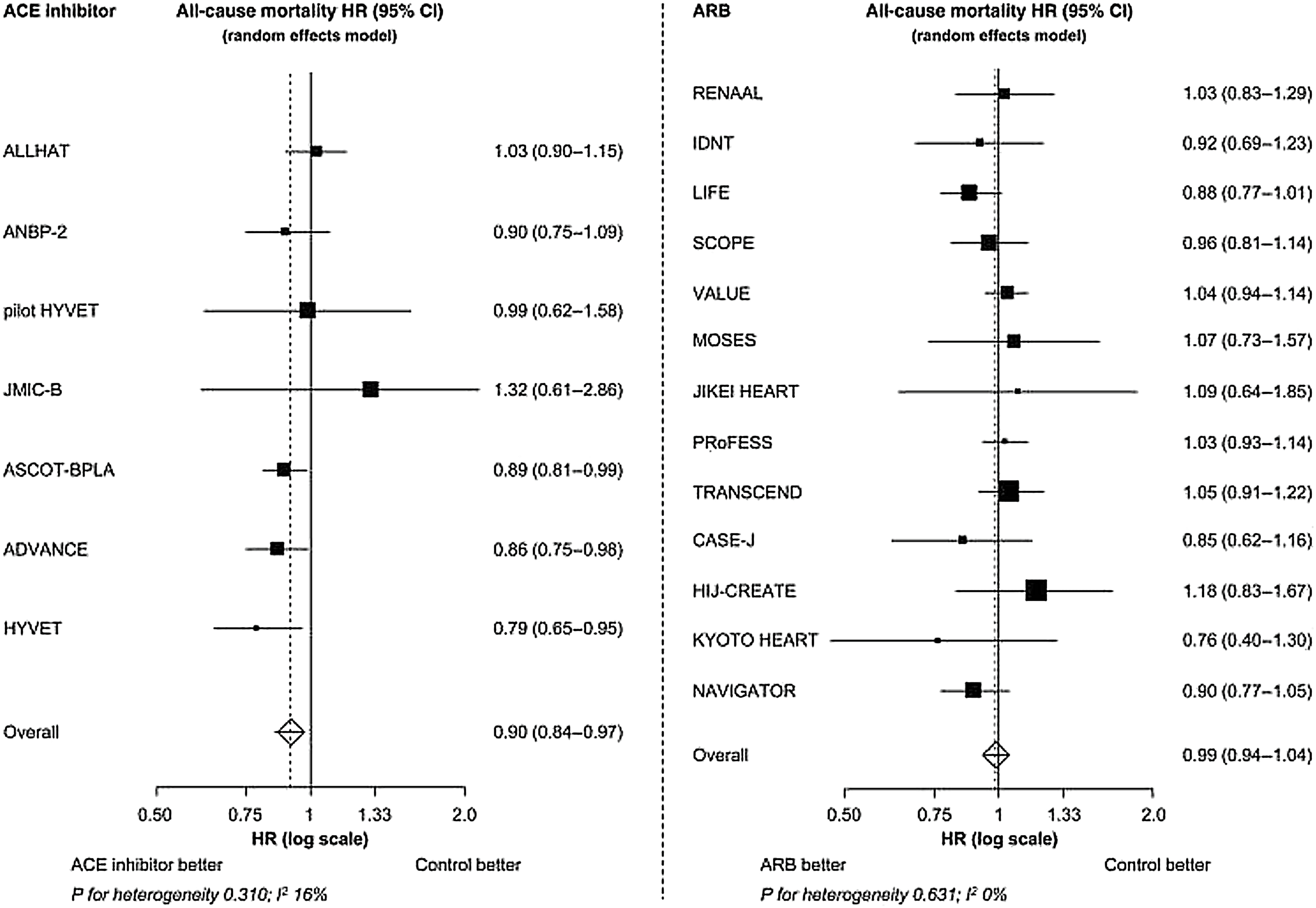
The evidence for the beneficial effects of ACEI on the cardiovascular outcomes was seen in several placebo-controlled studies ( Fig. 5.2 ). The Captopril Prevention Project (CAPPP) trial [ ] was a large-scale randomized controlled trial that compared the effects of ACEI and conventional therapy on the cardiovascular morbidity and mortality in patients with hypertension. More than 10,000 patients aged 25–66 years with a diastolic blood pressure >100 mmHg were randomized to captopril or diuretics and β-blockers. Both the treatment arms had similar reduction in blood pressure. For similar primary outcomes, captopril was associated with 25% reduced risk for stroke ( P = .04). In the Antihypertensive and Lipid-Lowering treatment to Prevent Heart Attack Trial (ALLHAT) [ ], more than 33,000 hypertensive subjects aged >55 years and at least one of other coronary heart disease risk factors were randomized to chlorthalidone, amlodipine, or lisinopril. Both amlodipine and chlortalidone arms achieved lower blood pressure than lisinopril arm with a high percentage of blood pressure goal <140/90 mmHg. After a 6-year follow-up, there was no difference in primary outcomes, while for the secondary outcomes the lisinopril arm showed a lower risk for combined cardiovascular risk, stroke, and heart failure. In a meta-analysis [ ] of 20 major randomized controlled trials on RAAS inhibitors in hypertension, ACEIs were associated with 10% reduction in all-cause ( P = .004), and 12% reduction in cardiovascular mortality ( P = .05). The meta-analysis included three trials [ ] that were not pure ACEI trials, and that accounted for the largest mortality reduction. Thus, the results should be interpreted with caution. Perindopril is an ACEI that is lipophilic and has a long duration of antihypertensive action [ , ]. In perindopril-based trials, compared to other compounds of the same class, in addition to reducing blood pressure, perindopril has been shown to have beneficial effect on endothelium [ ], regress atherosclerosis, and reduce arterial stiffness in patients with mild-to-moderate essential hypertension [ ].
ARBs
ARBs predominantly act by blocking the AT1R for Ang II and can be differentiated based on their affinity for this receptor. ARBs induce marked vasodilatation reducing both preload and afterload, thus, decreasing the systolic wall stress and left ventricular end-diastolic volume. The blockade of AT1R allows Ang II to exert its effect on AT2R which mediate the vasodilatation; improvement of vascular and cardiac function; and a reduction in cough from the absence of bradykinin increase. They also induce the activation of peroxisome proliferator-activated receptor-gamma and enhance the release of adiponectin from adipocytes which increase the insulin sensitivity, reduce the levels of circulating lipids, and promote antiinflammatory activity [ ]. In vitro and in vivo studies demonstrate that antiinflammatory effect of ARBs is through the suppression of the inflammatory toll-like receptors 2 (TLR2) which is also known as cluster of differentiation 282 (CD282) and TLR4, that are implicated in the development and progression of cardiovascular disease [ ]. In hypertensive patients, irbesartan has been shown to improve the endothelial function, vascular reactivity, and oxidative stress [ ]. Use of valsartan prevents the formation of reactive oxygen species and suppresses the activity of NF-κB that regulates the expression of inflammatory cytokines and cell adhesion molecules that contribute to vascular inflammation and vascular events [ ].
In hypertensive subjects, several trials have shown the efficacy of ARBs ( Fig. 5.2 ). In the Valsartan Antihypertensive Long-term Use Evaluation (VALUE) trial [ ], 15,245 patients with hypertension at high-risk for cardiovascular events aged >50 years were randomized to valsartan or amlodipine-based therapy. The mean BP reduction was 15/8 and 17/9 mmHg in the valsartan and amlodipine arms, respectively ( P < .0001 between groups). After a mean follow-up of 4.2 years, there was no significant difference in the primary outcome of cardiac mortality and morbidity. A subsequent meta-analysis [ ] found no increase in the risk of myocardial infarction while on treatment with ARBs. The Losartan Intervention For Endpoint reduction in hypertension (LIFE) trial [ ] evaluated 9193 hypertensive patients with left ventricular hypertrophy aged 55–80 years, treated with losartan or atenolol-based therapy. After a mean follow-up of 4.8 years, both treatments were associated with similar blood pressure reduction (−30/−17 mmHg), but the losartan group showed a 15% reduction in the primary outcome of cardiovascular mortality, stroke, and myocardial infarction ( P = .009), indicating benefits beyond blood pressure reduction. However, despite a reduction in blood pressure values, ARBs were not so effective in cardiovascular risk reduction compared to placebo in several trials [ ].
ARBs and ACEIs are recommended not only in the treatment of hypertension, but they have demonstrated to be effective in preventing the onset of microalbuminuria and to slow the progression toward renal failure [ ]. Their use is also recommended by international guidelines for patients with heart failure in view of their favorable effects on cardiovascular morbidity and mortality [ ]. With regards to the ischemic heart disease, some meta-analyses suggested that the ARBs are less effective than ACEIs, independent of blood pressure lowering effect, in preventing the myocardial infarction and all-cause mortality—the so called “ARB-MI Paradox” [ , ].
Direct renin inhibitors
Aliskiren is the first representative of the new class of orally active renin inhibitors [ ]. Comparable antihypertensive effects to most other agents were observed with aliskiren [ ]. The beneficial effects of aliskiren and its good tolerance in combinations with ARBs or a diuretic were confirmed in a meta-analysis on 12,000 patients with essential hypertension [ ]. Based on the results of the Aliskiren Trial in Type 2 Diabetes Using Cardiorenal Endpoints (ALTITUDE) [ ], with the increased incidence of adverse events (hyperkalemia and hypotension), co-administration of aliskiren with ACEIs or ARBs in patients with diabetes and moderate-to-advanced renal failure is not recommended. Recent analyses concerning renin inhibitors in their nephroprotection emphasize that double blockade of the RAAS can be considered in chronic nephropathy patients with albuminuria who are not fully controlled with conventional agents. In these patients, a strict monitoring of potassium levels and renal function should be stressed [ ].
Dual RAAS inhibition
The combination of an ACEI and ARB was initially considered to be a way of obtaining a stronger suppression of RAAS, which would improve the beneficial effects beyond blood pressure lowering. A meta-analysis of randomized controlled trials found that this combination reduced the ambulatory blood pressure by 4.7/3.0 mmHg compared with ACEI alone, and 3.8/2.9 mmHg compared with ARB alone [ ]. However, an increased risk of adverse events has been documented (worsening renal function, hyperkalemia, hypotension, and increased medical discontinuation due to side effects) [ ]. The additive effects of the dual blockade on blood pressure control have been shown to be less than that attained by either drug combined with a diuretic or a calcium channel blocker [ ]. The data from the ONTARGET study have shown that dual blockade of this type does not add any benefit in patients with high global cardiovascular risk and/or proteinuria, when compared to either agent used individually (monotherapy), and it increases the adverse events [ ].
ACEI versus ARBs
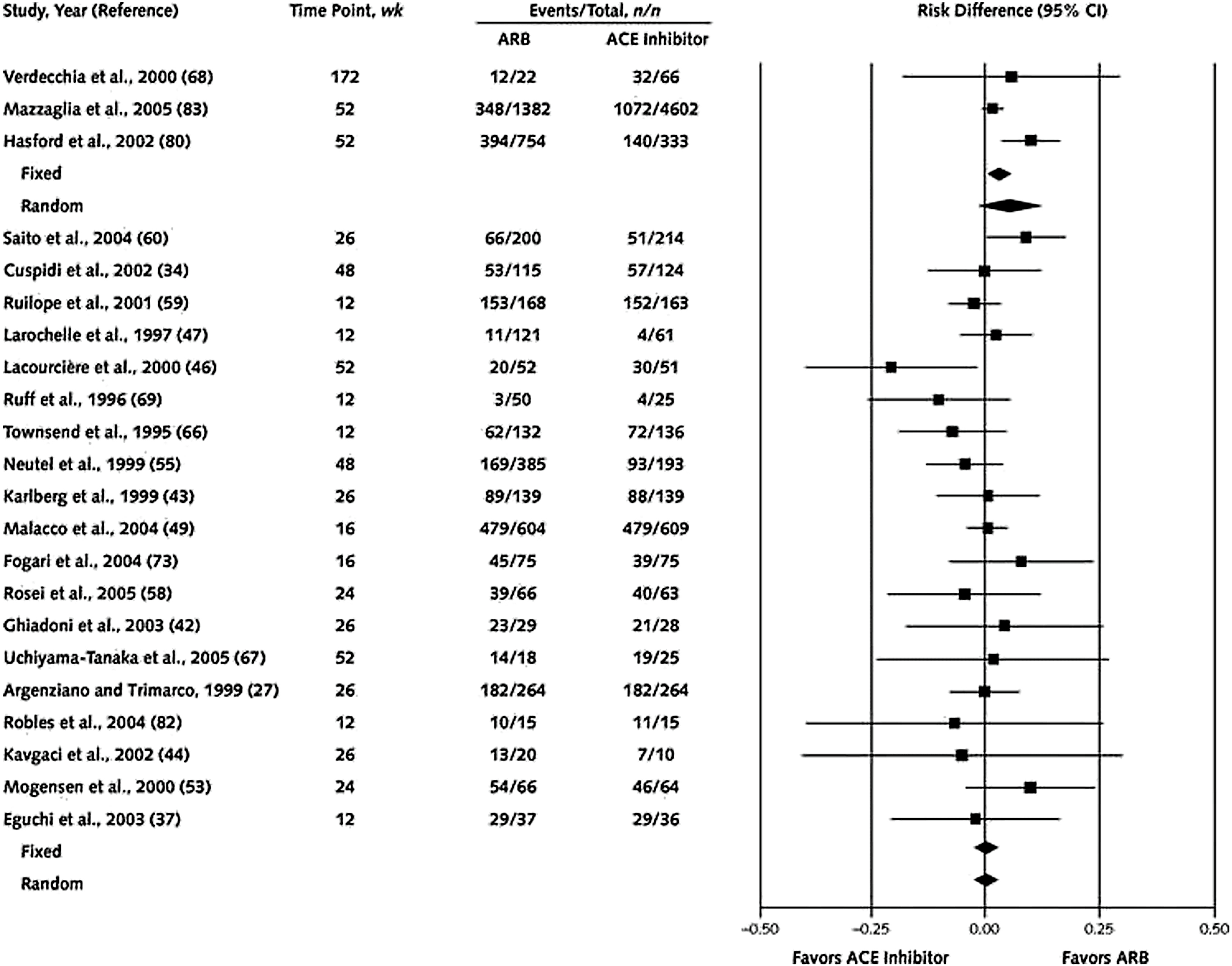
The antihypertensive efficacy of ACEIs and ARBS appears to be equivalent [ , ]. Across studies, the difference in systolic or diastolic blood pressure generally did not exceed 4 mmHg. There are limited trials that compared the cardiovascular outcomes between ACEIs and ARBs in hypertension [ ] ( Fig. 5.3 , [ ]). Other than ONTARGET [ ] previously described, there is another head-to-head comparison study known as the Diabetics Exposed to Telmisartan and Enalapril (DETAIL) study [ ]. In the DETAIL study, 250 type 2 diabetic subjects with early nephropathy and mild to moderate hypertension were randomized to receive either telmisartan or enalapril. After 5 years, there were no significant differences in the primary outcome of renal function and cardiovascular events, all-cause mortality, heart failure, or myocardial infarction. In both trials, ARBs reduced the blood pressure more than respective ACEI, and this could be due to the comparison of a long-acting ARB versus a short-acting ACEI administered once daily. As previously described, only in patients with ischemic heart disease, it has been shown that ARBs appear to be less effective than ACEIs, in preventing myocardial infarction and all-cause mortality independent of blood pressure lowering effect [ , ].