Regulation of Thyrotropin Secretion
Anthony N. Hollenberg
Thyrotropin (TSH) is the major regulator of thyroid hormone production. Measurement of serum TSH has become the principal screening test used by clinicians worldwide for the diagnosis of thyroid dysfunction. Thus, an understanding of the mechanisms by which TSH secretion is controlled is crucial for an understanding of normal thyroid physiology and for interpreting the results of measurements of serum TSH. In addition, regulation of TSH secretion by the thyrotroph cells of the pituitary is an ideal model in which to study the regulation of gene expression by thyroid hormones, thyrotropin-releasing hormone (TRH), and other substances. These regulators of TSH secretion will be considered individually, although it is likely that there are extensive interactions among them.
Anatomy
TSH is synthesized and secreted from the thyrotroph cells of the anterior pituitary and released into the circulation to regulate thyroid hormone synthesis in the thyroid (Fig. 10C.1). The thyrotrophs are exposed to circulating levels of thyroxine (T4) and triiodothyronine (T3), and these hormones, principally T3, tightly regulate the synthesis of TSH, mainly at the level of gene expression. In addition, the thyrotrophs are exposed to other hormones or drugs, such as glucocorticoids or retinoic acid analogs, which also may influence the synthesis and secretion of TSH. Besides the systemic circulation, the anterior pituitary receives direct communication from the hypothalamus via the capillaries that comprise the hypothalamic–pituitary portal system that arises from the median eminence. The most important hypothalamic regulator of TSH secretion is TRH. However, other hypothalamic neuropeptides and transmitters such as somatostatin and dopamine may also regulate TSH secretion. TRH neurons project from the paraventricular nucleus (located on either side of the third ventricle) to the median eminence, where TRH can be released and carried through the portal system to the pituitary (1,2).
The thyrotroph cells of the pituitary originate from a common pituitary stem cell and differentiate early in embryonic development (see Chapter 2). TRH is not required for the normal development of the thyrotrophs (3,4). The thyrotrophs share lineage with other pituitary cell types, including lactotrophs and somatotrophs, as demonstrated by mutations in the transcription factor POU1F1 (Pit-1), which lead to defects in all three cell types (5,6,7). In addition, the thyrotrophs share lineage with the gonadotroph cells, based on their shared expression of the common α-subunit of TSH, luteinizing hormone (LH), and follicle-stimulating hormone (FSH), and the key role of the transcription factor PROP1 in both gonadotroph and thyrotroph development in humans (8). It is clear that development of the thyrotrophs requires the action of several transcription factors including GATA2, PITX1 and 2, and LIM homeodomain transcription factors (9,10,11).
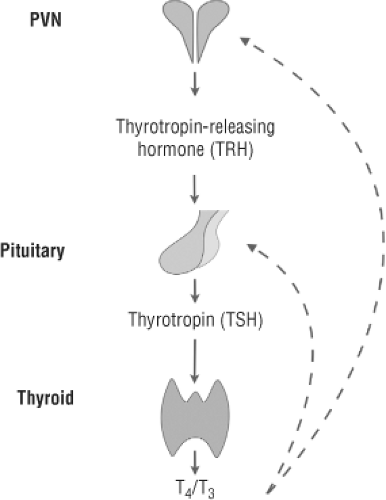 Figure 10C.1 Schematic diagram of the hypothalamic–pituitary–thyroid axis. TRH is synthesized in neurons within the paraventricular nucleus of the hypothalamus that project to the median eminence. At the median eminence, TRH is released into the hypothalamic–pituitary portal circulation and carried to the pituitary, where it activates TRH-1 receptors on the thyrotroph cells of the pituitary to synthesize and release TSH. TSH acts on the thyroid via TSH receptors to stimulate the synthesis and secretion of T4 and T3. T4 and T3 inhibit TRH and TSH secretion, closing the feedback loop. Courtesy of Ricardo De Costa Sousa, DVM Ph.D. |
Regulation of Thyrotropin Secretion
Like other pituitary hormones, TSH secretion is pulsatile, with 6 to 18 pulses per 24 hours in adult humans (12,13). The frequency and amplitude of the pulses are reasonably constant during the day, but there is an increase in both frequency and amplitude in the late evening and early morning (10 pm to 2 to 4 am) (14,15,16,17). As a result, serum TSH concentrations are about 2-fold higher during this period than during the rest of the day. This nocturnal surge of TSH secretion appears soon after birth, and is similar in males and females (18). The mechanisms
underlying TSH pulsatility and its nocturnal accentuation are not known, but likely include pulsatile TRH secretion and some input from the suprachiasmatic nucleus of the hypothalamus, which plays a role in many circadian rhythms in mammals (19). The pulsatility, especially at night, of TSH secretion decreases during fasting and nonthyroidal illness (20,21,22).
underlying TSH pulsatility and its nocturnal accentuation are not known, but likely include pulsatile TRH secretion and some input from the suprachiasmatic nucleus of the hypothalamus, which plays a role in many circadian rhythms in mammals (19). The pulsatility, especially at night, of TSH secretion decreases during fasting and nonthyroidal illness (20,21,22).
Thyrotropin-releasing Hormone
Production of TRH in the paraventricular nucleus of the hypothalamus is essential for normal TSH secretion. TRH-knockout mice have central hypothyroidism, with slightly high serum TSH concentrations, but the biologic activity of the TSH is decreased (23). Similarly, in humans TRH deficiency (secondary to structural hypothalamic lesions) results in a decrease in both the amount of TSH that is secreted and its biologic activity (24,25). Finally, mutations in the TRH receptor (TRH-R1) in the pituitary lead to central hypothyroidism in humans (26,27).
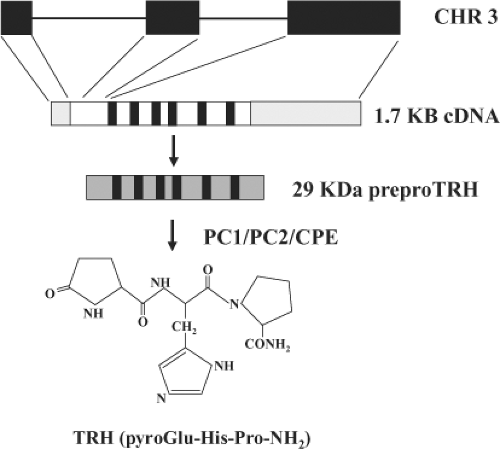 Figure 10C.2 Structures of the TRH gene, preproTRH, and TRH. The gene encoding human preproTRH is present on chromosome (CHR) 3 and is encoded by three exons. The first exon codes for the 5′-untranslated region and exons 2 and 3 code for the full 29-KDa preproTRH. The 3′-untranslated region is also coded for in exon 3. Human preproTRH contains six copies of the TRH progenitor sequence (black boxes), which are cleaved and further modified (see text) to yield mature TRH. PC denotes prohormone convertase; CPE, carboxypeptidase E. |
Structure and Synthesis of Thyrotropin-releasing Hormone
TRH is a tripeptide (pyroGlu-His-ProNH2) that is produced by the processing of a larger prohormone in the hypothalamus and elsewhere in the central nervous system (Fig. 10C.2) (1,28). It was the first hypothalamic hormone to be identified (29,30). The cDNA sequence of murine and rat TRH encodes for a preprohormone of approximately 29 kilodaltons (KDa), which includes five copies of the sequence of the TRH prohormone, whereas in humans the cDNA contains six copies (31,32,33). The hormone cDNA sequence is flanked by codons that encode pairs of basic amino acids that allow for processing of the progenitor TRH sequence (Gln-His-Pro-Gly) by two convertases, PC1 and PC2, and carboxypeptidase E (34,35). The progenitor TRH sequence is subsequently modified via cyclization at the N-terminus and amidation at the C-terminus to yield mature TRH (36,37).
The TRH mRNA also encodes several other peptides between the progenitor TRH sequences that can be isolated from the mammalian hypothalamus. TRH peptide 160–169, which lies between the third and fourth TRH progenitor sequences, has been isolated from rat hypothalamic tissue. This peptide can potentiate TRH-mediated TSH release from the anterior pituitary through a receptor separate from the TRH receptor that is probably located on non-endocrine cells of the pituitary (38,39,40). Rat TRH peptides 83–106 and 178–199 are both increased during suckling; they may contribute to prolactin release by blocking hypothalamic dopamine release (41). Several other pro-TRH-derived peptides have been isolated, including TRH-Gly, the direct precursor of TRH, but their role in vivo is not known (42). Related TRH peptides have also been identified in the human hypothalamus and placenta, but because the intervening sequences are different from that in rats, the sequences of these peptides are different (43,44), and their biologic activity, if any, is not known.
Based on the role of processing in producing mature TRH, it is clear that TRH production can be controlled through the regulation of enzymes such as PC-1 and PC-2 and carboxypeptidase E (2). Indeed, both PC-1 and PC-2 are downregulated by thyroid hormone and upregulated by leptin, consistent with their key regulatory role in the production of TRH (45,46,47). Mice that are deficient in carboxypeptidase E have less mature TRH and more of its precursor forms than normal mice (35). A human patient with defective PC-1 has been described; this patient had defective processing of several hormones, including insulin, and mild primary, not central, hypothyroidism (48,49).
TRH is rapidly deamidated after its release to TRH-free acid and histidyl-proline-diketopiperazine (His-Pro-DKP), a stable cyclized metabolite (37,50). The principal enzymes that degrade TRH are two forms of pyroglutamyl peptidase, PP 1 and PP II (also termed thyroliberase for the form produced peripherally). PP I is a cytosolic enzyme that has broad substrate specificity and has a role in the metabolism of other neuropeptides (50,51). In contrast, PP II while present in synaptosomal fractions of neural tissue and in pituitary cell membranes has recently been shown to be expressed in tanycytes, specialized glial cells that line the third ventricle and project to the median eminence where it can directly regulate TRH levels (52,53,54,55,56). Indeed, PP II has a substrate specificity restricted to TRH and TRH-like peptides, which, together with its tissue distribution, demonstrates that it is the major peptidase that removes the amino-terminal pyroglutamate residue from TRH (50,56,57). Furthermore, PP II levels can be regulated by T4 in tanycytes in vivo and may play a role in the rapid reduction of TSH after the administration of T4 (58).
The Thyrotropin-releasing Hormone Gene
The structure of the murine, rat, and human TRH genes are identical. Each consists of three exons and two introns (Fig. 10C.2). The sequences of the exons are well conserved among the species, whereas the sequences of the introns are less conserved. The first exon encodes the 5′-untranslated region,
while the coding sequence for the preprohormone and the 3′-untranslated region are on exons 2 and 3. The promoter is immediately adjacent to exon 1, as demonstrated by the presence of a TATA box within 25 base pairs (bp) of the transcription start site. In the hypothalamus, the content of preproTRH mRNA is highest in the paraventricular nucleus and the lateral hypothalamic area but it is also present in the dorsomedial and ventromedial hypothalamus (28,59). It is also found in the preoptic region, the olfactory lobes, the periaqueductal gray region, and the medullary raphe neurons in the brainstem, which provide much of the TRH found in the spinal cord. In addition, preproTRH mRNA is present in peripheral tissues, including the heart and the pancreas, where TRH may play a role in glucose homeostasis (23). In areas of the brain outside of the paraventricular nucleus, preproTRH mRNA and its derived peptides have several functions, including regulation of gastric motility and gastric acid secretion through activation of vagal outflow (60).
while the coding sequence for the preprohormone and the 3′-untranslated region are on exons 2 and 3. The promoter is immediately adjacent to exon 1, as demonstrated by the presence of a TATA box within 25 base pairs (bp) of the transcription start site. In the hypothalamus, the content of preproTRH mRNA is highest in the paraventricular nucleus and the lateral hypothalamic area but it is also present in the dorsomedial and ventromedial hypothalamus (28,59). It is also found in the preoptic region, the olfactory lobes, the periaqueductal gray region, and the medullary raphe neurons in the brainstem, which provide much of the TRH found in the spinal cord. In addition, preproTRH mRNA is present in peripheral tissues, including the heart and the pancreas, where TRH may play a role in glucose homeostasis (23). In areas of the brain outside of the paraventricular nucleus, preproTRH mRNA and its derived peptides have several functions, including regulation of gastric motility and gastric acid secretion through activation of vagal outflow (60).
The cell bodies of the TRH neurons (hypophysiotropic neurons) that control TSH secretion are located in the paraventricular nucleus. This nucleus has two major groups of neurons: A magnocellular group that is located laterally and expresses oxytocin and vasopressin, and a parvocellular group that is located medially. TRH neurons are found throughout the parvocellular group, but only those TRH neurons in certain anatomic subdivisions project to the median eminence to regulate thyrotroph function (1). The location of hypophysiotropic TRH neurons differs amongst species but are consistently found in the middle to dorsal PVN (59). The hypophysiotropic group of TRH neurons are also defined by their co-expression of the mRNA encoding the cocaine- and amphetamine-related neuropeptide (CART) (61,62) and by their ability to regulate preproTRH mRNA levels as discussed below. The role of TRH neurons in the paraventricular nucleus that do not express CART and do not project to the median eminence is not clear. A separate group of medial parvocellular neurons of the paraventricular nucleus contain corticotropin-releasing hormone, the key hypothalamic regulator of corticotropin (ACTH) secretion. Corticotropin-releasing hormone is also present in other areas of the paraventricular nucleus during stress (63).
The underlying mechanism allowing for cell-specific TRH gene expression is not known. However, the transcription factor simple-minded (Sim)-1 is required for the formation of all paraventricular-nucleus neurons in vivo, and mice that lack Sim-1 have little development of the paraventricular nucleus and the supraoptic nucleus of the hypothalamus (64,65,66). There is also a role for the related transcription factor Sim-2 in the development of TRH neurons in the PVN (67). It is likely that Sim-1 and its obligate partner, the transcription factor aryl hydrocarbon receptor nuclear translocator (Arnt)-2 act upstream of Sim-2 and potentially another transcription factor (Brn-2) to allow appropriate development of these two hypothalamic nuclei (68,69). In addition, the transcription factor Orthopedia (Otp) is also critical for the development of the paraventricular nucleus; it acts before Sim-1 in the developing hypothalamus (70,71). The factors that cause the development of separate TRH neurons from other neuropeptide-expressing neurons in the paraventricular nucleus are not known, but it is likely that still unidentified cell-specific factors are needed for the development of these neurons, perhaps by interacting directly with regulatory elements in the TRH promoter which are required for its hypothalamic expression (72).
Regulation of the Expression of the Thyrotropin-releasing Hormone Gene
TRH gene expression is dynamically regulated in hypophysiotropic neurons of the PVN to regulate TSH secretion and subsequently thyroid secretion in response to many conditions, including illness, starvation, cold, and thyroid disease. An understanding of the mechanisms involved can be discerned through an examination of both the milieu in which the hypophysiotropic TRH neurons lie and the regulatory elements present within the TRH gene.
The TRH neurons in the paraventricular nucleus receive input from other regions of the brain as well as from the circulation. The major afferent connections to these neurons include catecholamine neurons from the brain stem (73) and neurons from the arcuate nucleus (1,74). Catecholamine signaling likely plays an important role in the upregulation of TRH gene expression during cold exposure (75), while input from the arcuate nucleus plays a critical role in the downregulation of TRH gene expression during starvation. Indeed, insight into the regulation of TRH neurons by inputs from the arcuate nucleus has shed considerable light on the importance of pathways via which energy expenditure regulates the axis.
In addition to neuronal connections, TRH neurons in the PVN are also dependent upon glial cells for delivery of key effectors such as triiodothyronine (T3). As discussed previously, specialized glial cells termed tanycytes surround the third ventricle and play a key role in the regulation of TRH. These tanycytes, in addition to PPII, express the type 2 deiodinase (dio2) and appear to be responsible for the local production of T3 in the PVN (76,77). The expression of dio2 in these tanycytes is upregulated by inflammatory signaling pathways which suggests that enhanced local production of T3 could be, in part, responsible for the suppression of TRH in the euthyroid sick syndrome (78). However, it still is not clear how the T3 produced in tanycytes accesses the PVN.
Regulation by Pathways Important in Energy Expenditure
In rodents, fasting results is a rapid fall in serum TSH and thyroid hormone concentrations (79,80), caused, in part, by downregulation of TRH gene expression in the paraventricular nucleus (81,82). Administration of the adipocyte hormone leptin, the production of which also falls during fasting, prevents the fasting-induced fall in thyroid secretion by preventing the fall in TRH mRNA levels (83,84). Thus, leptin is a key regulator of TRH gene expression (Fig. 10C.3). The actions of leptin in the hypothalamus are mediated by the long-form of the leptin receptor (Ob-Rb), a member of the cytokine receptor family that is coupled to the Janus-kinase 2 – signal transducer of activated transcription 3 (JAK2-STAT3) signaling cascade (85). The Ob-Rb receptor is expressed strongly in the arcuate nucleus and in a much more limited way in the paraventricular nucleus (86,87,88), and is thus poised to regulate TRH expression both indirectly and directly (89,90,91). Lesions of the arcuate nucleus prevent the effects of leptin on TRH gene expression (92).
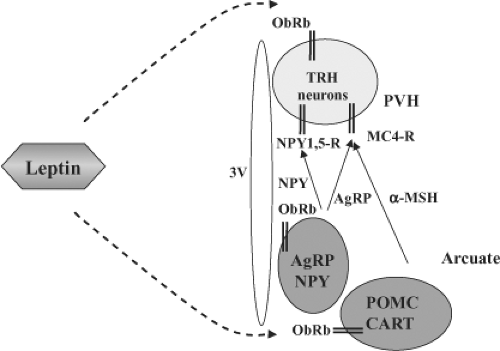 Figure 10C.3 Schematic diagram of the actions of leptin on TRH gene expression. Leptin, produced in adipocytes, crosses the blood–brain barrier and regulates TRH gene expression either directly via leptin receptors (ObRB) on TRH neurons or indirectly via the arcuate nucleus. The arcuate nucleus contains at least two neuronal populations that have leptin receptors and project to TRH neurons to regulate TRH expression: (1) POMC neurons synthesize α-MSH that stimulates TRH expression through the MC4-R. (2) AgRP/NPY neurons produce both AgRP and NPY, which inhibit TRH expression. AgRP is an inverse agonist of the MC4-R, while NPY signals through its own receptors on TRH neurons (NPY 1,5-R). AgRP denotes Agouti-related protein; CART, cocaine- and amphetamine-related neuropeptide; α-MSH, α-melanocyte-stimulating hormone; MC4-R, melanocortin-4 receptors; NPY, neuropeptide Y; NPY1,5-R, neuropeptide-1 and -5 receptors; ObRb, leptin receptor; POMC, pro-opiomelanocortin; PVH, paraventricular nucleus; 3V, third ventricle. |
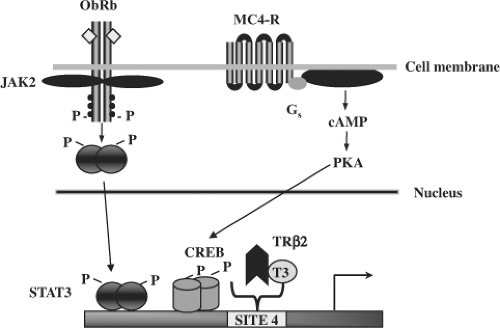 Figure 10C.4 Activation of the TRH promoter by multiple signaling pathways. The TRH promoter responds to leptin, melanocortin, and T3 via a number of cis-acting elements. Binding of leptin to its receptor (ObRb) activates JAK2, leading to its phosphorylation and subsequent recruitment and phosphorylation (P) of STAT3. Phosphorylated STAT3 homodimers enter the nucleus and bind directly to the promoter region of the TRH gene to stimulate its expression. The POMC (pro-opiomelanocortin)-derived peptide, α-MSH (melanocyte-stimulating hormone), activates TRH transcription via PKA (protein kinase A) by binding to the MC4-R (melanocortin-4 receptor) on TRH neurons. PKA phosphorylates CREB (cyclic-AMP response element–binding protein) bound to the TRH promoter, which leads to TRH transcription. In contrast, T3 binds to the β2 isoform of TR (thyroid receptor), which acts through Site 4 to downregulate TRH gene expression. JAK2 denotes Janus-kinase 2, and STAT3, signal transducer of activated transcription 3; together they from the JAK2-STAT3 signaling cascade. |
Two neuronal groups within the arcuate nucleus express leptin receptors and project to the TRH neurons in the paraventricular nucleus. The neurons in the first group contain both pro-opiomelanocortin (POMC) and CART (93,94,95). In the arcuate nucleus, unlike the pituitary, POMC is processed primarily to α-melanocyte-stimulating hormone (α-MSH),
a potent anorexigenic peptide, which signals through both the melanocortin (MC) 3 and 4 receptors. TRH neurons in the paraventricular nucleus have MC4 receptors and are directly innervated by α-MSH nerve terminals (96,97). In addition, central administration of α-MSH prevents the fasting-induced suppression of TRH (89,90). The second group of arcuate-nucleus neurons, which have leptin receptors, also synthesize the neuropeptides agouti-related peptide (AgRP) and neuropeptide Y. These neurons also directly contact TRH neurons in the paraventricular nucleus. In contrast to POMC, both AgRP and NPY are orexigenic peptides and are downregulated by leptin and upregulated during fasting (98,99,100). Both AgRP, a MC4-receptor antagonist (and an inverse agonist), and NPY (through NPY-receptor isoforms expressed on TRH neurons), when administered centrally, can cause central hypothyroidism by downregulating TRH mRNA expression in the paraventricular nucleus (101,102,103,104). In vivo, the expression of NPY appears to be absolutely required for the suppression of TRH during a fast. Remarkably, though, even if TRH suppression is prevented during a fast in mice that lack NPY, thyroid hormone levels still fall as enhanced hepatic metabolism occurs through a pathway that is also controlled by NPY and MC4R signaling (105). In summary, the arcuate nucleus serves to integrate leptin signaling and assist in the regulation of TRH gene expression as well as regulate food intake and energy expenditure (106,107).
a potent anorexigenic peptide, which signals through both the melanocortin (MC) 3 and 4 receptors. TRH neurons in the paraventricular nucleus have MC4 receptors and are directly innervated by α-MSH nerve terminals (96,97). In addition, central administration of α-MSH prevents the fasting-induced suppression of TRH (89,90). The second group of arcuate-nucleus neurons, which have leptin receptors, also synthesize the neuropeptides agouti-related peptide (AgRP) and neuropeptide Y. These neurons also directly contact TRH neurons in the paraventricular nucleus. In contrast to POMC, both AgRP and NPY are orexigenic peptides and are downregulated by leptin and upregulated during fasting (98,99,100). Both AgRP, a MC4-receptor antagonist (and an inverse agonist), and NPY (through NPY-receptor isoforms expressed on TRH neurons), when administered centrally, can cause central hypothyroidism by downregulating TRH mRNA expression in the paraventricular nucleus (101,102,103,104). In vivo, the expression of NPY appears to be absolutely required for the suppression of TRH during a fast. Remarkably, though, even if TRH suppression is prevented during a fast in mice that lack NPY, thyroid hormone levels still fall as enhanced hepatic metabolism occurs through a pathway that is also controlled by NPY and MC4R signaling (105). In summary, the arcuate nucleus serves to integrate leptin signaling and assist in the regulation of TRH gene expression as well as regulate food intake and energy expenditure (106,107).
The direct and indirect actions of leptin on TRH neurons to regulate TSH and thyroid secretion in situations of nutritional stress are probably mediated, in part, by changes in TRH gene transcription. The proximal regions of the promoter regions of the rodent and human TRH genes are structurally similar (Fig. 10C.4). Both of the genes have a TATA box within 25 bp of the transcriptional start site and a region termed Site 4 between 60 and 52 bp upstream of the start site. First identified as a thyroid hormone receptor–binding site (see later) (32,108), Site 4 is critical for basal activity of the TRH promoter in mammalian cell lines. Though Site 4 can bind the cyclic-AMP response element–binding protein (CREB), which is downstream in the signaling cascade induced by α-MSH via MC4 receptors, another site in the TRH promoter (-101 to -94) with higher affinity for CREB has recently been identified (109,110). Thus, modulation of CREB activity via α-MSH, AgRP, and NPY signaling should allow for varying degrees of TRH gene expression. The TRH gene also has a conserved STAT-binding site between 150 and 140 bp upstream of the start site. This site interacts with STAT3 and mediates transcriptional responses to leptin signaling via the ObRb receptor (74,111,112). Thus, the promoter region of the TRH gene has sites whereby multiple inputs can be integrated to control TRH gene expression.
While most of the studies of nutritional regulation of TRH production, or at least gene expression, have been performed in rodents, there is increasing evidence that similar mechanisms affect TRH production and therefore TSH and thyroid secretion in humans during food deprivation and illness. Humans with mutations of the leptin receptor have central hypothyroidism, although humans who have a defective leptin gene
do not (113). Furthermore, controlled caloric restriction in humans leading to weight loss results in a decline in serum thyroid hormone concentrations that can be reversed by leptin (114,115). Also, acute fasting in humans leads to a decrease in pulsatile TSH secretion; this decrease also can be reversed by the administration of leptin (20,22). However, humans with MC4-receptor mutations, the most common genetic form of obesity, have normal serum TSH and thyroid hormone concentrations (116,117).
do not (113). Furthermore, controlled caloric restriction in humans leading to weight loss results in a decline in serum thyroid hormone concentrations that can be reversed by leptin (114,115). Also, acute fasting in humans leads to a decrease in pulsatile TSH secretion; this decrease also can be reversed by the administration of leptin (20,22). However, humans with MC4-receptor mutations, the most common genetic form of obesity, have normal serum TSH and thyroid hormone concentrations (116,117).
Regulation of Expression of the Thyrotropin-releasing Hormone Gene by Nonthyroidal Illness
Many changes in pituitary–thyroid function occur in nonthyroidal illness, including a decrease in TSH secretion (see section on nonthyroidal illness in Chapter 11C). One cause of the decrease in TSH secretion is a decrease in TRH secretion, as manifested by a decrease in TRH gene expression in the paraventricular nucleus (21,118). The frequent presence of anorexia in these patients suggests that the leptin and melanocortin signaling pathways may play a role in the decrease. However, it is also likely that cytokines such as interleukin-1 and -1β, tumor necrosis factor, and interferon contribute to the decrease. MC4-R knockout mice do not develop cachexia in response to cytokine administration or an increased tumor burden, suggesting that the melanocortin signaling pathway plays an important role in the response. However, increased expression of arcuate-nucleus neuropeptides during cachexia does not by itself explain low TRH gene expression or, for that matter, decreased food intake. It is likely that interactions between the cytokine-signaling pathways and the melanocortin system contribute to the central suppression of TRH gene expression in the paraventricular nucleus during nonthyroidal illness, or that other cytokine-activated pathways are involved (119,120,121). Furthermore, as outlined, increased expression of dio2 in tanycytes appears to increase local concentrations of T3 in the PVN and lead to TRH suppression. Indeed, mouse models that lack dio2 do not suppress TRH expression in response to lipopolysaccharide (LPS), whereas LPS leads to suppression of TRH in wild-type mice (122).
Regulation of Thyrotropin-releasing Hormone Gene Expression by Thyroid Hormone
An important component of the control of TSH secretion is inhibition of TRH gene expression in the paraventricular nucleus by thyroid hormone, principally T3. Conversely, TRH gene expression increases in response to low serum T4 and T3 concentrations (123,124). TRH mRNA levels in other regions of the hypothalamus are not regulated by T3. In terms of serum hormones, T4 may be more important than T3 (125), with the T3 being produced locally from T4 by the action of dio2 which is expressed in tanycytes as described previously. This is supported by the finding that D2-knockout mice maintain TSH secretion in the presence of high serum T4 concentrations (77,126,127). Once T3 is produced locally in the hypothalamus by tanycytes, next it must gain access to TRH neurons. While it is now known that the transport of T3 across the blood–brain barrier requires the monocarboxylate 8 transporter (MCT8), it is also clear that the MCT8 is also present on TRH neurons in the PVN (128,129,130). Further work will be required to understand how local T3 from tanycytes accesses the TRH neurons in the PVN via the MCT8 (131).
T3 regulates TRH gene expression at the level of transcription via its nuclear receptor, the thyroid hormone receptor (TR) (Fig. 10C.4) (132). There are three TR isoforms, α1, β1, and β2, which bind T3 and mediate transcriptional regulation (see Chapter 8). The TRβ isoforms are alternatively spliced products of a single gene and differ structurally only in the amino terminus, while TRα1 is the product of a separate gene but functions in the same way as the TRβ isoforms (133,134). The greatest difference in each of the isoforms may be in their cell-specific expression patterns (135). Mice devoid of both TRβ isoforms have high serum TSH and high T4 and T3 concentrations, findings that resemble those found in patients with resistance to thyroid hormone (see Chapter 58), consistent with the predominant expression of TRβ isoforms in the pituitary and hypothalamus (136,137). The values are similar in mice with selective ablation of the β2 isoform, demonstrating the requirement for this isoform for negative regulation by T3 of TRH gene expression (138). This isoform is also required for the normal development of color vision due to its unique expression in the retina (139). It is also possible that β1 and α1 isoforms play a role in the regulation of TRH gene expression (140,141,142).
Related posts:
 Anatomy and Pathology of the Thyrotrophs
Anatomy and Pathology of the Thyrotrophs
 Iodine Deficiency and Endemic Cretinism
Iodine Deficiency and Endemic Cretinism
 Toxic Adenoma and Toxic Multinodular Goiter
Toxic Adenoma and Toxic Multinodular Goiter
 Thyroid Hormones in Thermogenesis, Intermediary Metabolism, and Obesity
Thyroid Hormones in Thermogenesis, Intermediary Metabolism, and Obesity
 Overview of the Clinical Manifestations of Hypothyroidism
Overview of the Clinical Manifestations of Hypothyroidism
 Hypothyroidism in Infants and Children: Neonatal Screening
Hypothyroidism in Infants and Children: Neonatal Screening
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


