Reduced Sensitivity to Thyroid Hormone: Defects of Transport, Metabolism, and Action
Alexandra M. Dumitrescu
Samuel Refetoff
Resistance to thyroid hormone (RTH), a syndrome of reduced responsiveness of target tissues to thyroid hormone (TH) was identified in 1967 (1). An early report proposed various mechanisms including defects in TH transport, metabolism, and action (2). However, with the identification of TH receptor (TR) β gene mutations 22 years later (3,4), the term RTH become synonymous with defects of this gene (5). Subsequent discoveries of genetic defects that reduce the effectiveness of TH through altered cell membrane transport (6,7) and metabolism (8) have broadened the definition of TH hyposensitivity to encompass all defects that can interfere with the biologic activity of a chemically intact hormone secreted in normal or even excess amounts. In this revised chapter, we have retained the acronym RTH to denote the syndrome produced by reduced intracellular action of the active TH, T3. The term of reduced sensitivity to TH (RSTH) is used to denote reduced effectiveness of TH in the broader sense.
TH Secretion, Cell-Membrane Transport, Metabolism, and Action
Proper TH action requires 1) an intact TH, 2) its transport across cell membrane, 3) hormone activation through intracellular metabolism, 4) cytosolic processing and nuclear translocation, 5) binding to receptors and 6) interaction with co-regulators or other post receptor effects mediating the TH effect.
Maintenance of TH supply is insured by a feedback control mechanism involving the hypothalamus, pituitary, and thyroid gland (see Fig. 58.1A). A decrease in the circulating TH concentration induces a hypothalamus-mediated stimulation of TSH secretion from the pituitary thyrotrophs, which stimulates the thyroid follicular cells to synthesize and secrete more hormones. In contrast, TH excess shuts down the system through the same pathway to reinstate homeostasis. This centrally regulated system does not respond to changing requirements for TH in a particular organ or cell.
Additional systems operate to accommodate for local TH requirements. One such system is the control of TH entry into the cell through active transmembrane transporters (9). Another is the activation of the hormone precursor thyroxine (T4) by removal of the outer ring iodine (5′-deiodination) to form triiodothyronine (T3) or, inactivate T4 and T3 by removal of the inner ring iodine (5-deiodination) to form reverse T3 (rT3) and T2, respectively (see Fig. 58.1B). Cell specific adjustment in deiodinase activity allows for additional local regulation of hormone supply (10).
Finally, the types and abundance of TRs, through which TH action is mediated, determine the nature and degree of the response. TH action takes place in the cytosol as well as in the nucleus (11). The latter, known as genomic effect, has been more extensively studied (12,13) (see Fig. 58.1C). TRs are transcription factors that bind to DNA of genes whose expression they regulate.
 Figure 58.1 Regulation of TH supply, metabolism and genomic action. A: Feedback control that regulates the amount of TH in blood. B: Intracellular metabolism of TH, regulating TH bioactivity. C: Genomic action of TH. For details see text. CBP/P300, cAMP-binding protein/general transcription adaptor; TFIIA and TFIIB, transcription intermediary factor II, A and B; TBP, TATA-binding protein; TAF, TBP-associated factor. |
How Thyroid Hormone Deficiency and Excess Coexist
TH deficiency and excess are associated with typical symptoms and signs reflecting the global effects of lack and excess of the hormone, respectively, on all body tissues. A departure from this became apparent with the identification of the RTH syndrome. Subjects with RTH have high TH levels without TSH suppression. This paradox encompasses other biochemical and clinical observations, suggesting TH deficiency, sufficiency, and excess, depending on the degree and nature of the TR abnormality (5). The syndrome of TH cell-membrane transport defect (THCMTD) presents a similar paradox, as subjects have high serum T3 concentrations but the uptake of TH is not uniform in all tissues and cell types (14).
Resistance to Thyroid Hormone (Rth)
In practice, patients with RTH are identified by their persistent elevation of circulating free TH levels in association with non-suppressed serum TSH, and in the absence of intercurrent illness, drugs, or alterations of TH transport serum proteins. More importantly, higher doses of exogenous TH are required to produce the expected suppressive effect on the secretion of pituitary TSH and the expected metabolic responses in peripheral tissues.
Although the apparent resistance to TH may vary in severity, it is always partial. The variability in clinical manifestations may be due to the severity of the hormonal resistance, the effectiveness of compensatory mechanisms, the presence of modulating genetic factors, and the effects of prior therapy.
The magnitude of the hormonal resistance is, in turn, dependent on the nature of the underlying genetic defect. The latter is usually, but not always, a mutation in the TRβ gene (5,15).
The magnitude of the hormonal resistance is, in turn, dependent on the nature of the underlying genetic defect. The latter is usually, but not always, a mutation in the TRβ gene (5,15).
Despite a variable clinical presentation, the common features characteristic of the RTH syndrome are: 1) elevated serum levels of free T4 and to a lesser degree T3, particularly in older individuals, 2) normal or slightly increased TSH level that responds to TRH, 3) absence of the usual symptoms and metabolic consequences of TH excess, and 4) goiter.
Clinical Classification
The diagnosis is based on the clinical findings and standard laboratory tests and confirmed by genetic studies. Before TRβ gene defects were recognized, the proposed sub-classification of RTH was based on symptoms, signs and laboratory parameters of tissue responses to TH (16). Notwithstanding the assessment of TSH feedback regulation by TH, the measurements of most other responses to the hormone are insensitive and relatively nonspecific. For this reason, all tissues other than the pituitary have been grouped together under the term peripheral tissues, on which the impact of TH was roughly assessed by a combination of clinical observation and laboratory tests.
The majority of patients appeared to be eumetabolic and maintained a near normal serum TSH concentration. They were classified as having generalized resistance to TH (GRTH). In such individuals, the defect seemed to be compensated by the high levels of TH. In contrast, patients with equally high levels of TH and nonsuppressed TSH that appeared to be hypermetabolic, because they were restless or had sinus tachycardia, were classified as having selective pituitary resistance to TH (PRTH). Finally, the occurrence of isolated peripheral tissue resistance to TH (PTRTH) was reported in a single patient (17). No mutation in the TRβ gene of this patient was found (18) and no similar cases have been reported. Thus, it is uncertain whether PTRTH exists as a true thyrotroph specific TH resistance entity, except in association with TSH-producing pituitary adenomas caused by expression of somatic mutation or isoform specific TRβs (19,20). More common in clinical practice is the apparent tolerance of some individuals to the ingestion of supraphysiologic doses of TH.
Table 58.1 Types of TRβ Gene Mutations | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The earliest suggestion that PRTH may not constitute an entity distinct from GRTH can be found in a study by Beck-Peccoz et al. (21). A comprehensive study involving 312 patients with GRTH and 72 patients with PRTH, has conclusively shown that the response of SHBG and other peripheral tissue markers of TH action, were equally attenuated in GRTH
and PRTH (22). More importantly, identical mutations were found in individuals classified as having GRTH and PRTH, many of whom belonged to the same family (23). It was, therefore, concluded that these two forms of RTH are the product of the subjective nature of symptoms as well as the individual’s target organ susceptibility to changes of TH, also observed in subjects with thyroid dysfunction in the absence of RTH (see section on the Molecular Basis of the Defect).
and PRTH (22). More importantly, identical mutations were found in individuals classified as having GRTH and PRTH, many of whom belonged to the same family (23). It was, therefore, concluded that these two forms of RTH are the product of the subjective nature of symptoms as well as the individual’s target organ susceptibility to changes of TH, also observed in subjects with thyroid dysfunction in the absence of RTH (see section on the Molecular Basis of the Defect).
Incidence and Inheritance
The precise incidence of RTH is unknown. Because routine neonatal screening programs are based on the determination of TSH, RTH is rarely identified by this means (24). A limited neonatal survey by measuring blood T4 concentration, suggested the occurrence of one case per 40,000 live births (25,26). Known cases surpass 3,000.
Although most thyroid diseases occur more commonly in women, RTH has been found with equal frequency in both genders. The condition appears to have wide geographic distribution and has been reported in Caucasians, Africans, Asians and Amerindians. The prevalence may vary among different ethnic groups.
Familial occurrence of RTH has been documented in approximately 75% of cases. Taking into account only those families in whom both parents of the affected subjects have been studied, the true incidence of sporadic cases, is 21.0%. This is in agreement with current estimate of the frequency of de novo mutations of 20.8% (see Table 58.1). The reports of acquired RTH are seriously questioned.
Inheritance is autosomal dominant. Transmission was clearly recessive in only one family (1,27). Consanguinity in three families with dominant inheritance of RTH has produced homozygous children with very severe clinical manifestations (28,29).
 Figure 58.2 Location of natural mutations in the TRβ molecule associated with RTH. TOP PORTION: Schematic representation of the TRβ and its functional domains for interaction with TREs (DNA-binding) and with hormone (T3-binding). Their relationship to the three clusters of natural mutations is also indicated. TRβ2 has 15 more residues than TRβ1 at the aminoterminus. BOTTOM PORTION: The location of the 170 different mutations detected and their frequencies in the total of 457 unrelated families (published and our unpublished data). Amino acids are numbered consecutively starting at the amino terminus of the TRβ1 molecule according to the consensus statement of the First International Workshop on RTH (31). “Cold regions” are areas devoid of mutations associated with RTH. |
Etiology and Genetics
Using the technique of restriction fragment length polymorphism, Usala et al. (30) were first to demonstrate linkage between a TRβ locus on chromosome 3 and the RTH phenotype. Subsequent studies at the University of Chicago and
at the National Institutes of Health have identified distinct point mutations in the TRβ gene of two unrelated families with RTH (3,4). In both families only one of the two TRβ alleles was involved, compatible with the apparent dominant mode of inheritance.
at the National Institutes of Health have identified distinct point mutations in the TRβ gene of two unrelated families with RTH (3,4). In both families only one of the two TRβ alleles was involved, compatible with the apparent dominant mode of inheritance.
Mutations in the TRβ gene have now been identified in subjects with RTH belonging to 457 families (see Table 58.1 and Fig. 58.2). They comprise 170 different mutations. With the exception of the index family, found to have complete deletion of the TRβ gene (27), the majority (430 families) have single nucleotide substitutions resulting in single amino acid replacements in 419 instances and stop codons in 11 others, producing truncated molecules. In addition, deletions, insertions and a duplication were identified in 20 families (for details see Table 58.1).
Given that there are 287 more families than the 170 different mutations, 78 of the mutations are shared by more than one family. Haplotyping of intragenic polymorphic markers showed that, in most instances, identical mutations have developed independently in different families (32). These occur more often, though not exclusively, in CpG dinucleotide hot spots. In fact, de novo mutations are twice as frequent in CpG dinucleotides. In addition, different mutations producing more than one amino acid substitution at the same codon have been found at 44 different sites. Mutations in codons 345 and 451 each produced 5 different amino acid replacements (G345R, S, A, V, D; F451I, L, S, C, X) while those in codon 453, seven (P453T, S, A, N, Y, H, L) not counting an insertion and a deletion. A total of 59 families harbor mutations at codon 453. Mutations are located in the last four exons of the gene: 6, 17, 73, and 73 mutations in exons 7, 8, 9, and 10, respectively. These involve 35, 23, 202, and 196 families (see Fig. 58.2). The following mutations have been identified in more than 15 families: R243Q, A317T, R338W, R423H and P453T. Of note the
first three are in CpG dinucleotides and the last in a stretch of six cytidines. Thirty-three unrelated families share the R338W mutation.
first three are in CpG dinucleotides and the last in a stretch of six cytidines. Thirty-three unrelated families share the R338W mutation.
All TRβ gene mutations are located in the functionally relevant domain of T3-binding and its adjacent hinge region. Three mutational clusters have been identified with intervening cold regions (see Fig. 58.2). With the exception of the family with TRβ gene deletion, inheritance is autosomal dominant in all others.
Somatic mutations in the TRβ gene have been identified in some TSH-secreting pituitary tumors (19,33). These mutations can be identical to those occurring in the germline. However, because their expression is limited to the thyrotrophs, the phenotype, as in other TSHomas, is that of TSH induced thyrotoxicosis. It is postulated that defective TR interfering with the negative regulation of TSH by TH is responsible for the development of the pituitary tumor.
In 14% of families, RTH occurs in the absence of mutations in the TR genes (nonTR-RTH) (34). Such individuals may have a defect in one of the cofactors involved in the mediation of TH action (see Animal Models of RTH, below).
Molecular Basis of the Defect
Thyroid Hormone Action
TH receptor genes located on chromosome 17 and 3, generate a TRα and a TRβ molecule, respectively, with substantial structural and sequence similarities. Both genes produce two isoforms; α1 and α2 by alternative splicing and β1 and β2 by different transcription start points. TRα2 binds to TH response elements (TREs) but due to a sequence difference at the ligand-binding domain (LBD) site, it does not bind TH (35) and appears to have a weak antagonistic effect (36). Additional TR isoforms, including a TRβ with shorter amino terminus (TRβ3), truncated TRβ3, TRα1 and TRα2, lacking the DNA-binding domain (DBD), have been identified in rodents (37,38) and TRβ4 that lacks the LBD in selected human tissues (39). Their significance in humans remains unknown (40). Finally, a p43 protein, translated from a downstream AUG of TRα1, is believed to mediate the TH effect in mitochondria (41).
The relative expression of the two TR genes and the distribution of their products vary among tissues and during different stages of development (42,43,44). The abundance of several splice variants involving the 5′-untranslated region of the human TRβ1 (45,46) is developmentally and tissue regulated. Although TRβ and TRα are interchangeable (47,48) to a certain degree, the absence of one or the other receptor do not produce equivalent phenotypes. Some TH effects are absolutely TR isoform specific (see Animal Models of RTH, below).
TREs, located in TH regulated genes, consist of half-sites having the consensus sequence of AGGTCA and vary in number, spacing and orientation (49,50). Each half-site usually binds a single TR molecule (monomer) and two half-sites bind two TRs (dimer) or one TR and a heterologous partner (heterodimer), the most prominent being the retinoid X receptor γ (RXR). Dimer formation is facilitated by the presence of an intact “leucine zipper” motif located in the middle of the LBD of TRs. Occupation of TREs by unliganded (without hormone) TRs, also known as aporeceptors, inhibits the constitutive expression of genes that are positively regulated by TH (51) through association with corepressors such as the nuclear corepressor (NCoR) or the silencing mediator of retinoic acid and TH receptors (SMRT) (52). Transcriptional repression is mediated through the recruitment of the mammalian homologue of the Saccaromyces transcriptional corepressor (mSin3A) and histone deacetylases (HDAC) (53). This latter activity compacts nucleosomes into a tight and inaccessible structure, effectively shutting down gene expression (see Fig. 58.1C). This effect is relieved by the addition of TH, which releases the corepressor, reduces the binding of TR dimers to TRE, enhances the occupation of TREs by TR/RXR heterodimers (54) and recruits coactivators (CoA) such as p/CAF (CREB binding protein-associated factor) and nuclear coactivators (NCoA) (55) with HAT (histone acetylation) activity (52,56). This results in the loosening of the nucleosome structure making the DNA more accessible to transcription factors (see Fig. 58.1C). Actually, the ligand-dependent association with TR associated proteins, in conjunction with the general coactivators PC2 and PC4, act to mediate transcription by RNA polymerase II and general initiation factors (57). Furthermore, it is believed that T3 exerts its effect by inducing conformational changes of the TR molecule and that TR associated proteins (TRAP) stabilizes the association of TR with TRE.
In addition to the genomic effect described above, TH acts at the cell membrane and cytosol (11). These non-genomic effects include oxidative phosphorylation and mitochondrial gene transcription and involve the generation of intracellular secondary messengers with induction of [Ca(2+)](I), cyclic adinosine monophosphate (cAMP) AMP or protein kinase signaling cascades.
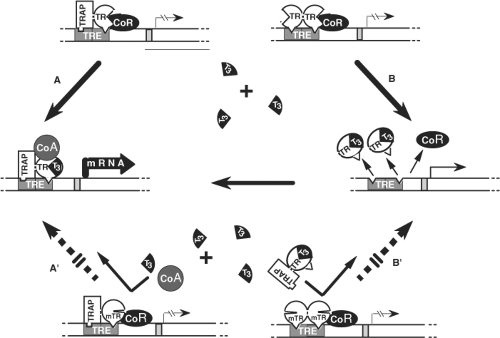 Figure 58.3 Mechanism of the dominant expression of RTH: In the absence of T3, occupancy of TRE by TR heterodimers (TR-TRAP) or dimers (TR–TR) suppresses transactivation through association with a corepressor (CoR). A: T3-activated transcription mediated by TR-TRAP heterodimers involves the release of the CoR and association with coactivators (CoA) as well as B: the removal of TR dimers from TRE releases their silencing effect and liberates TREs for the binding of active TR-TRAP heterodimers. The dominant negative effect of a mutant TR (mTR), that does not bind T3, can be explained by the inhibitory effect of mTR-containing-dimers and heterodimers that occupy TRE. Thus, T3 is unable to activate the mTR-TRAP heterodimer (A′) or release TREs from the inactive mTR homodimers (B′). (Modified from Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocr Rev 1993;14:348–399.) |
Properties of Mutant TRβ Receptors and Dominant Negative Effect
TRβ gene mutations produce two forms of RTH. The less common, described in only one family (1), is caused by deletion of all coding sequences of the TRβ gene and is inherited as an autosomal recessive trait (27). The complete lack of TRβ in these individuals produces severe deafness, resulting in mutism (1), as well as monochromatic vision (58) as TRβ is required for the cochlear maturation and the development of cone photoreceptors that mediate color vision (59) (see Animal Models of RTH, below). Heterozygous individuals that express a single TRβ gene have no clinical or laboratory abnormalities. This is niether due to compensatory overexpression of the single normal allele of the TRβ gene nor that of the TRα gene (60). However, because subjects with complete TRβ gene deletion preserve some TH responsiveness, it is logical to conclude that TRα1 is capable of partially substituting for the function of TRβ (see Animal Models of RTH, below).
The more common form of RTH is inherited in a dominant fashion and is characterized by defects in one allele of the TRβ gene, principally missense mutations. This contrasts with the lack of phenotype in individuals that express a single TRβ allele. These mutant TRβs (mTRs) do not act by reducing the amount of a functional TR (haploinsufficiency) but by interfering with the function of the wild-type (WT) TR (dominant negative effect, DNE). This has been clearly demonstrated
in experiments in which mTRs are coexpressed with WT TRs (61,62).
in experiments in which mTRs are coexpressed with WT TRs (61,62).
Studies have established two basic requirements for mTRs to exert a DNE: (1) preservation of binding to TREs on DNA and (2) the ability to dimerize with a homologous (63,64) or heterologous (65,66) partner. These criteria apply to mTRs with predominantly impaired T3-binding activity (see Fig. 58.3). In addition, a DNE can be exerted through impaired association with a cofactor even in the absence of important impairment of T3-binding. Increased affinity of an mTR for a corepressor (CoR) (67,68), or reduced association with a coactivator (CoA) (69,70,71), have been found to play a role in the dominant expression of RTH. The introduction in an mTR of an additional artificial mutation that abolishes either DNA-binding, dimerization or the association with a CoR results in the abrogation of its DNE (66,72,73).
The distribution of TRβ gene mutations associated with RTH reveals conspicuous absence of mutations in regions of the molecule that are important for dimerization, for the binding to DNA and for the interaction with CoR (see Fig. 58.2). These “cold regions” contain CpG hot spots, suggesting that they may not be devoid of natural mutations. Rather, mutations would escape detection owing to their failure to produce clinically significant RTH in heterozygotes, as tested in vitro (74). Structural studies of the DBD and LBD have provided further understanding about the clustered distribution of mTRβs associated RTH and defects in the association with cofactors (75,76,77,78).
On the basis of the early finding that RTH is associated with mutations confined to the LBD of the TRβ, it was anticipated that the clinical severity of RTH would correlate with the degree of T3-binding impairment. While this was true in 12 different natural mTRβs, in five others, the severity of RTH was lesser despite virtually complete absence of T3-binding. This was explained by the reduced dominant negative potency due to diminished ability to form homodimers (for example R316H and R338W) (79). Weakened association of TRβ with DNA or CoR can produce the same effect.
Less evident was the observation of relatively severe interference with the function of the WT TRβ, despite very mild impairment or no T3-binding defect at all. This was the case when hormone-binding was tested in two mTRβs, located in the hinge region of the receptor (R243Q and R243W) (80). However, reduced T3-binding could be demonstrated after complexing to TRE, indicating a change in the mTRβ configuration when bound to T3 (80,81). Other mechanisms and examples of DNE in the presence of normal or slightly attenuated T3-binding are: decreased interaction of L454Vwith the CoA (69) and delay of R383H to release the CoR (82).
In general the relative degree of impaired function among various mTRβs is similar whether tested using TREs controlled reporter genes that are negatively or positively regulated by T3.
Exceptions to this rule are the mTRβs, R383H, and R429Q that show greater impairment of transactivation on negatively rather than positively regulated promoters (79,82,83). In this respect these two mTRβs are candidates for predominantly PRTH, even though they have been clinically described as producing GRTH (84) as well as PRTH (85,86). Recent work suggests that the substitution of these charged aminoacids (here arginines) disrupts the unique property of TRβ2 to bind certain coactivators through multiple contact surfaces (87). The result is a decrease in the normal T3-mediated feedback suppression by converting the TRβ2 to a TRβ1-like single mode of coactivator binding. As a consequence, the mutation affects predominantly TRβ2 mediated action. In vivo support for a TRβ2 predominant impairment of the mTRβ R429Q was obtained in mice (88). Another possible mechanism for PRTH is a double-hit combining a single nucleotide polymorphism (SNP) and the mTRβ R338W (89). The presence of a thymidine in a SNP, located in the enhancer region of the TRβ gene, leads to over-expression of the mutant allele in GH3 pituitary-derived cells. However, the T/C nucleotides of this SNP have not been correlated with the clinical presentation in individuals with this most common TRβ R338W mutation.
Exceptions to this rule are the mTRβs, R383H, and R429Q that show greater impairment of transactivation on negatively rather than positively regulated promoters (79,82,83). In this respect these two mTRβs are candidates for predominantly PRTH, even though they have been clinically described as producing GRTH (84) as well as PRTH (85,86). Recent work suggests that the substitution of these charged aminoacids (here arginines) disrupts the unique property of TRβ2 to bind certain coactivators through multiple contact surfaces (87). The result is a decrease in the normal T3-mediated feedback suppression by converting the TRβ2 to a TRβ1-like single mode of coactivator binding. As a consequence, the mutation affects predominantly TRβ2 mediated action. In vivo support for a TRβ2 predominant impairment of the mTRβ R429Q was obtained in mice (88). Another possible mechanism for PRTH is a double-hit combining a single nucleotide polymorphism (SNP) and the mTRβ R338W (89). The presence of a thymidine in a SNP, located in the enhancer region of the TRβ gene, leads to over-expression of the mutant allele in GH3 pituitary-derived cells. However, the T/C nucleotides of this SNP have not been correlated with the clinical presentation in individuals with this most common TRβ R338W mutation.
Molecular Basis of the Variable Phenotype of RTH
The extremes of the RTH phenotype have a clear molecular basis. Subjects heterozygous for a TRβ gene deletion are normal because the expression of a single TRβ allele is sufficient for normal function. RTH manifests in homozygotes completely lacking the TRβ gene and in heterozygotes that express an mTRβ with DNE. The most severe form of RTH, with extremely high TH levels and signs of both hypothyroidism and thyrotoxicosis, occur in homozygous individuals expressing only mTRβs (28,29). The severe hypothyroidism manifesting in bone and brain of such subjects can be explained by the silencing effect of a double dose of mTR and its interference with the function of TRα (63); a situation which does not occur in homozygous subjects with TRβ deletion. In contrast, the manifestation of thyrotoxicosis in other tissues, such as the heart, may be explained by the effect high TH levels have on tissues that normally express predominantly TRα1 (90,91) (see Animal Models of RTH, below). It is for this same reason that tachycardia is a relatively common finding in RTH (92).
Various mechanisms can be postulated to explain the tissue differences in TH resistance within the same subject and among individuals. The distribution of receptor isoforms varies from tissue to tissue (42,93,94). This likely accounts for greater hormonal resistance of the liver as compared to the heart. Differences in the degree of resistance among individuals harboring the same mTRβ could be explained by the relative level of mutant and WT TR expression. Such differences have been found in one study using cultured fibroblasts (95) but not in another (60). Various reasons for a predominant TRβ2 dysfunction have been presented in the preceding section.
Although in a subset of mTRβs a correlation was found between their functional impairment and the degree of thyrotroph hyposensitivity to TH, this correlation was not maintained with regards to the hormonal resistance of peripheral tissues (79). Subjects with the same mutations, even belonging to the same family, show different degrees of RTH. A most striking example is that of family G.H. in which the mTRβ R316H did not cosegregate with the RTH phenotype in all family members (96). This variability of clinical and laboratory findings was not observed in affected members of two other families with the same mutation (23,97). A study in a large family with the mTRβ R320H, suggests that genetic variability of factors other than TR may modulate the phenotype of RTH (98).
RTH Without Tr Gene Mutations (nonTR-RTH)
The molecular basis of nonTR-RTH remains unknown. Since the first demonstration of nonTR-RTH (15), 49 subjects belonging to 39 different families have been identified (34,99,100). The phenotype is indistinguishable from that in subjects harboring TRβ gene mutations (see differential diagnosis, below). Distinct features are an increased female to male ratio of 3.5:1 and the high prevalence of sporadic cases. As a matter of fact, of the 35 families in which both parents, all siblings and progeny were examined, the occurrence of RTH in another family member was documented in only 6. In those instances, and as in the case of TRβ-linked RTH, the inheritance pattern is dominant. While it has been postulated that nonTR-RTH is likely caused by a defect in one of the cofactors involved in the mediation of TH action, proof supporting this contention is lacking (101).
Animal models of RTH
Understanding the action of TH in vivo, and the mechanisms underlying the abnormalities observed in patients with RTH, has been bolstered by observations made in genetically manipulated mice. Three types of genetic manipulations have been applied: (a) transgenic mice that over express a receptor; (b) deletion of the receptor (knockout or KO); and (c) introduction of mutations in the receptor (knockin or KI). The latter two types of gene manipulation, species differences not withstanding, have yielded true models of the recessively and dominantly inherited forms of RTH, respectively (102).
The features of RTH found in patients homozygous for TRβ deletion also manifest in the TRβ deficient mouse (103,104,105). Special features, such as sensorineural deafness and monochromatic vision are characteristic and shared by mouse (106,107) and man (1,58). The mouse model allowed for investigations in greater depth into the mechanisms responsible for the development of these abnormalities. Thus, TRβ deficiency retards the expression of fast-activating potassium conductance in inner hair cells of the cochlea that transforms the immature cells from spiking pacemakers to high-frequency signal transmitters (108). TRβ2 interacts with transcription factors providing timed and spatial order for cone differentiation. Its absence results in the selective loss of M-opsin (107). The down regulation of hypothalamic TRH is also TRβ2 specific (109). Mice deficient in TRβ have increased heart rate that can be decreased to the level of the WT mouse by reduction on the TH level (105). This finding, together with the lower heart rate in mice selectively deficient in TRα1 (90), indicates that TH dependent changes in heart rate are mediated through TRα, and explains the tachycardia observed in some patients with RTH.
The combined deletion of TRα1 and α2, produces no important alterations in TH or TSH concentrations in serum (47). The complete lack of TRs, both α and β, is compatible with life (47,48). This contrasts with the complete lack of TH which, in the athyreotic Pax8 deficient mouse, results in death before weaning, unless rescued by TH treatment (110). The survival of mice deficient in both TRα and β is not due to expression of a yet unidentified TR but to the absence of the noxious effect of aporeceptors. Indeed, removal of the TRα gene rescues the Pax8 KO mice from death (111). The combined TRβ and TRα deficient mice have serum TSH levels that are 500-fold higher than those of the WT mice, and T4 concentrations 12-fold above the average normal mean (47). Thus, the presence of an aporeceptor does not seem to be required for the upregulation of TSH but any amount of TH causes its downregulation.
The first animal model of the organ-limited dominant RTH utilized somatic transfer of an mTRβ1 G345R gene by means of a recombinant adenovirus (112). The liver of these mice was resistant to TH, and overexpression of the WT TRβ increased the severity of hypothyroidism, confirming that the unliganded TR has a constitutive effect in vivo as in vitro. True mice models of dominantly inherited RTH have been generated by targeted mutations in the TRβ gene (113,114). Mutations were modeled on those identified in humans with RTH [frame-shift resulting in 16 carboxyl-terminal nonsense amino acids (PV mouse) and T337Δ]. As in humans, the phenotype manifested in the heterozygous KI animals and manifestations were more severe in the homozygotes.
NcoA (SRC-1) deficient mice have RTH with typical increase in T4, T3, and TSH concentrations (115). A more mild form of RTH was identified in mice deficient in RXRγ (116). Animals show reduced sensitivity to L-T3 in terms of TSH downregulation but not in metabolic rate. These data indicate that abnormalities in cofactors can produce RTH. The significance and mechanism of the hypotalamo-pituitary-thyroid activation in the Jun N-terminal kinase 1 (Jnk1) KO mouse has not been yet determined (117).
The Phenotype of TRα Gene Mutation in Mouse and Humans
The question of why mutations in the TRα gene have not been identified earlier in man was partially answered by the study of mice with targeted gene manipulations. As stated in the preceding section, TRα gene deletions, total or only α1, failed to produce a RTH phenotype. Similarly, mice with targeted TRα gene mutations failed to manifest the phenotype of RTH. Several human mutations known to occur in the TRβ gene were targeted in homologous regions of the TRα gene of the mouse. These are, the PV frame-shift mutation, TRα1 R384C (equivalent to TRβ R438C) in the and TRα P398H (equivalent to TRβ P452H) and TRa L400R (corresponding to TRβ454) (118). While the resulting phenotypes were somewhat variable, none exhibited thyroid tests abnormalities characteristic of RTH. A common feature in heterozygotes was retarded post-natal development and growth, decreased heart rate, and difficulty in reproducing. Also, all were lethal in the homozygous state, in accordance with the noxious effect of unliganded TRα1.
The recent identification of a TRα gene mutation in a human recapitulates the findings in the TRα KI mice (119). This nonsense mutation produces a truncated TRα1 (E403X) that lacks the C-terminal α-helix. As a consequence, in addition to a negligible T3-binding, the mutation promotes corepressor binding while abolishing binding of the coactivator, both contributing to a strong DNE. The 6-year-old girl, harboring this mutation, presented with chronic constipation noted upon weaning at 7 months of age, and growth and developmental delay. Hypothyroidism manifested in organs expressing predominantly the TRα, including bone, gastrointestinal tract, heart, striated muscle, and central nervous system. More specifically X-rays showed patent cranial sutures with wormian bones, delayed dentition, femoral epiphyseal dysgenesis and retarded bone age. In addition diminished colonic motility with megacolon, slow heart rate, reduced muscle strength were suggestive of hypothyroidism, as was her placid affect, slow monotonous speech and cognitive impairment. Thyroid function tests showed, as in the mouse with truncated TRα, a low serum T4, high T3, and very low rT3, somewhat reminiscent of MCT8 defects (see the THCMTD Section in this chapter), presumably due to alterations in iodothyronine metabolism.
Pathogenesis
The reduced sensitivity to TH in subjects with RTH is shared to a variable extent by all tissues. The hyposensitivity of the pituitary thyrotrophs results in nonsuppressed serum TSH, which in turn, increases the synthesis and secretion of TH. The persistence of TSH secretion in the face of high levels of free TH contrasts with the low TSH levels in the more common forms of TH hypersecretion that are TSH-independent. This apparent paradoxical dissociation between TH and TSH is responsible for the wide use of the term “inappropriate secretion of TSH” to designate the syndrome. However, TSH hypersecretion is not at all inappropriate, given the fact that the response to TH is reduced. It is compensatory and appropriate for the level of TH action mediated through a defective TR. As a consequence most patients are eumetabolic, though the compensation is variable among affected individuals and among tissues in the same individual. However, the level of tissue responses do not correlate with the level of TH, probably owing to a discordance between the hormonal effect on the pituitary and other body tissues. Thyroid gland enlargement occurs with chronic, though minimal TSH hypersecretion due to increased biologic potency of this glycoprotein through increased sialylation (120). Administration of supraphysiologic doses of TH is required to suppress TSH secretion without induction of thyrotoxic changes in peripheral tissues.
Thyroid-stimulating antibodies, which are responsible for the thyroid gland hyperactivity in Graves’ disease, have been conspicuously absent in patients with RTH. Another potential thyroid stimulator, human chorionic gonadotropin, has not been found in serum of subjects with RTH (121,122).
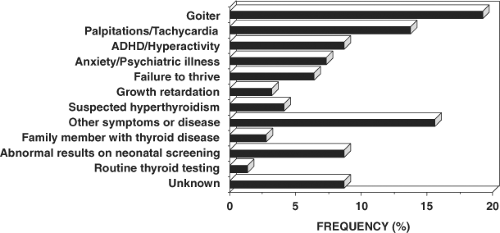 Figure 58.4 The reasons prompting further investigation of the index member of each family with RTH. |
The selectivity of the resistance to TH has been convincingly demonstrated. When tested at the pituitary level, both thyrotrophs and lactotrophs were less sensitive only to TH. Thyrotrophs responded normally to the suppressive effects of the dopaminergic drugs L-dopa and bromocriptine (123,124) as well as to glucocorticoids (124,125,126). Studies carried out in cultured fibroblasts confirm the in vivo findings of selective resistance to TH. The responsiveness to dexamethasone, measured in terms of glycosaminoglycan (127) and
fibronectin synthesis (128), was preserved in the presence of T3 insensitivity.
fibronectin synthesis (128), was preserved in the presence of T3 insensitivity.
Several of the clinical features encountered in some patients with RTH may be the manifestation of selective tissue deprivation of TH during early stages of development. These clinical features include retarded bone age, stunted growth, mental retardation or learning disability, emotional disturbances, attention deficit/hyperactivity disorder (ADHD), hearing defects, and nystagmus (5). A variety of associated somatic abnormalities appear to be unrelated pathogenically and may be the result of involvement of other genes such as in major deletions of DNA sequences (27). However, no gross chromosomal abnormalities have been detected on karyotyping (1,129).
Pathology
Little can be said about the pathologic findings in tissues other than the thyroid. Electron microscopic examination of striated muscle obtained by biopsy from one patient revealed mitochondrial swelling, also known to be encountered in thyrotoxicosis (1). This is compatible with the predominant expression of TRα in muscle, responding to the excessive amount of circulating TH (130). Light microscopy of skin fibroblasts stained with toluidine blue showed moderate to intense metachromasia (2) as described in myxedema. However, in contrast to patients with TH deficiency, treatment with the hormone failed to induce the disappearance of the metachromasia in fibroblasts from patients with RTH.
Thyroid tissue, obtained by biopsy or at surgery, revealed various degrees of hyperplasia of the follicular epithelium (124,131,132,133). Specimens have been described as “adenomatous goiters”, “colloid goiters” and normal thyroid tissue. When present, lymphocytic infiltration is due to the coexistence of thyroiditis (134).
Table 58.2 Clinical Features | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Clinical Features
Characteristic of the RTH syndrome is the paucity of specific clinical manifestations. When present, manifestations are variable from one patient to another. Investigations leading to the diagnosis of RTH have been undertaken because of the presence of goiter, hyperactive behavior or learning disabilities, developmental delay and sinus tachycardia (see Fig. 58.4). The finding of elevated serum TH levels in association with nonsuppressed TSH is usually responsible for the pursuit of further studies leading to the diagnosis.
The degree of compensation to tissue hyposensitivity by the high levels of TH is variable among individuals as well as in different tissues. As a consequence, clinical and laboratory evidence of TH deficiency and excess often coexist. For example, RTH can present with a mild-to-moderate growth retardation, delayed bone maturation and learning disabilities suggestive of hypothyroidism, alongside with hyperactivity and tachycardia compatible with thyrotoxicosis. The more common clinical features and their frequency are given in Table 58.2. Frank symptoms of hypothyroidism are more common in those individuals who, because of erroneous diagnosis, have received treatment to normalize their circulating TH levels.
Goiter is by far the most common abnormality. It has been reported in 66% to 95% of cases and is almost always detected by ultrasonography. Gland enlargement is usually diffuse; nodular changes and gross asymmetry are found in recurrent goiters after surgery.
Sinus tachycardia is also very common, with some studies reporting frequency as high as 80% (22). Palpitations often bring the patient to the physician and the finding of tachycardia is the most common reason for the erroneous diagnosis of autoimmune thyrotoxicosis or the suspicion of PRTH.
About one-half of subjects with RTH have some degree of learning disability with or without ADHD (5,135). One-quarter have intellectual quotients (IQ) less than 85% but frank mental retardation (IQ <60) has been found only in 3% of cases. Impaired mental function was found to be associated with impaired or delayed growth (<fifth percentile) in 20% of subjects though growth retardation alone is rare (4%) (5). Despite the high prevalence of ADHD in patients with RTH, the occurrence of RTH in children with ADHD must be very rare, none having been detected in 330 such children studied (136,137). The higher prevalence of low IQ scores appear to confer a higher likelihood for subjects with RTH to exhibit ADHD symptoms (97). A retrospective survey has shown an increased miscarriage rate and low birth weight of normal infants born to mothers with RTH (138).
A variety of physical defects that cannot be explained on the basis of TH deprivation or excess have been recorded. These include major or minor somatic defects, such as winged scapulae, vertebral anomalies, pigeon breast, prominent pectoralis, birdlike facies, scaphocephaly, craniosynostosis, short fourth metacarpals, as well as Besnier’s prurigo, congenital ichthyosis, and bull’s eye type macular atrophy (5). Some may be related to the severity of the hormonal resistance manifested in homozygotes (29).
Course of the Disease
The course of the disease is as variable as is its presentation. Most subjects have normal growth and development, and lead a normal life at the expense of high TH levels and a small goiter. Others present variable degrees of mental and growth retardation. Symptoms of hyperactivity tend to improve with age as it does in subjects with ADHD only.
Goiter has recurred in every patient who underwent thyroid surgery. As a consequence, some subjects have been submitted to several consecutive thyroidectomies or treatments with radioiodide (133,139,140,141).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


