Central Hypothyroidism
Luca Persani
Paolo Beck-Peccoz
Introduction
Central hypothyroidism (CH) is a rare disease characterized by a defect of thyroid hormone production due to an insufficient stimulation by thyrotropin (TSH) of an otherwise normal thyroid gland (1). This condition can be the consequence of an anatomic or functional disorder of the pituitary gland or the hypothalamus. Because these disorders frequently affect both sites and defective TSH secretion is the common final result, the formerly used terms, “secondary hypothyroidism of pituitary origin,” and “tertiary hypothyroidism of hypothalamic origin” resulting from insufficient TSH stimulation by thyrotropin-releasing hormone (TRH), are used less frequently. CH is a heterogeneous disease due to the wide spectrum of mechanisms accounting for its pathogenesis. Indeed, CH can be congenital in the case of genetic defects or acquired in the case of lesions, such as tumors, traumas, or vascular accidents, affecting the hypothalamic–pituitary region. In some cases, CH is an isolated defect of pituitary function, but in most cases it is combined with other pituitary hormone deficiencies (CPHDs). In many cases, the hypothyroid state is less severe than expected and its manifestations are frequently masked by concomitant pituitary defects. Diagnosis is usually made biochemically with low circulating free T4 (FT4) concentrations associated with low/normal serum TSH levels. Treatment of CH is by administration of levothyroxine (LT4). Follow-up treatment cannot be easily evaluated as in primary hypothyroidism since circulating TSH has lost much of its significance as a measure of thyroid hormone action in central defects. Therefore, the only measurement useful for monitoring the correct substitution dose of LT4 is the circulating free thyroid hormone concentration.
Epidemiology
Hypothyroidism is the most prevalent endocrine dysfunction, but CH is rare and accounts for a small portion of hypothyroid patients. CH prevalence in the general population is estimated to range from 1:20,000 to 1:80,000 (2). Neonatal screening programs for hypothyroidism based on combined TSH/T4-TBG tests indicate that the presence of CH ranges from 1:16,000 to 1:100,000 live newborns (3,4). Most of these cases are genetic forms of CPHDs. CH can affect patients of all ages and, different from what is observed in primary hypothyroidism, the female to male ratio is around 1.0. CH itself does not severely reduce life expectancy, though the quality of life can be severely affected by the hypothyroid state. Therefore, the existence of mild forms of CH should always be suspected in patients with hypothalamic–pituitary disorders.
Table 38.1 Possible Causes of Central Hypothyroidism (CH) | |
|---|---|
|
Table 38.2 Genetic Forms of Central Hypothyroidism (CH) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Pathogenesis
The pathogenesis of CH is quite heterogeneous and the disease mechanisms are still largely unexplained (1,5). The
mechanisms leading to the defect in TSH secretion variably involve both hypothalamic and pituitary cells. Depending on the underlying organic or genetic causes, sporadic or familial forms of CH have been described. The major causes of CH are listed in Table 38.1 and that for genetic forms of CH with their phenotypic manifestations is provided in Table 38.2. In the vast majority of both congenital and acquired forms of CH, the thyrotrope defect is combined with other pituitary hormone deficiencies. Indeed, isolated CH is mainly due to specific genetic defects, such as TSH beta subunit (TSHβ) and TRH receptor gene mutations.
mechanisms leading to the defect in TSH secretion variably involve both hypothalamic and pituitary cells. Depending on the underlying organic or genetic causes, sporadic or familial forms of CH have been described. The major causes of CH are listed in Table 38.1 and that for genetic forms of CH with their phenotypic manifestations is provided in Table 38.2. In the vast majority of both congenital and acquired forms of CH, the thyrotrope defect is combined with other pituitary hormone deficiencies. Indeed, isolated CH is mainly due to specific genetic defects, such as TSH beta subunit (TSHβ) and TRH receptor gene mutations.
 Figure 38.1 Serum levels of free T4 and immunoreactive TSH in a large series of patients with central hypothyroidism. The lack of correlation is consistent with a reduced bioactivity of circulating TSH molecules. (Modified from Persani L, Ferretti E, Borgato S, et al. Circulating TSH bioactivity in sporadic central hypothyroidism. J Clin Endocrinol Metab 2000;85:3631–3635.) |
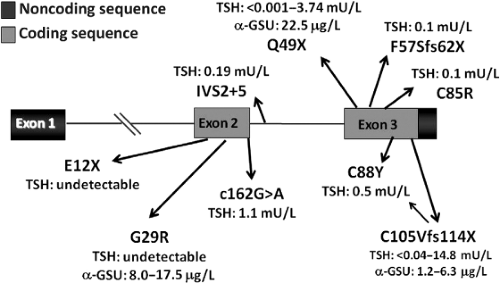 Figure 38.2 Schematic representation of TSHβ gene with illustration of the known mutations. Homozygosity was seen in all affected cases except one who was compound heterozygous for C88Y and C105Vfs114X, configuring a typical recessive disease. Serum TSH and glycoprotein hormone α-subunit (α-GSU) circulating levels observed in the affected patients are reported. Note that TSH levels were, in some instances, in the normal range, whereas α-GSU was always elevated indicating the defective dimerization with mutant TSHβ subunit. |
Defects in TSH secretion may be quantitative (the so-called reduced “thyrotropin reserve”) or qualitative, or both (6,7,8,9,10,11,12,13). In genetic CH forms, the defect is usually quantitative. Indeed, serum TSH is undetectable/low in most cases of genetic CH, a typical example being TSHβ gene mutations that result in the synthesis of a truncated TSHβ subunit often unable to dimerize with glycoprotein hormone α-subunit (α-GSU). In contrast, the defect is frequently both quantitative and qualitative in acquired CH. The quantitative defect in TSH producing cells is frequently associated with a qualitative defect in the secreted TSH isoforms that conserve immunoreactivity, but display a severe impairment in intrinsic biological activity and ability to stimulate TSH receptors. The secretion of bioinactive TSH has been reported in CH due to hypothalamic–pituitary tumors or breech delivery (8,9,10,11), external radiation for head tumors (12) and Sheehan’s syndrome (13). The existence of this qualitative defect in TSH secretion provides an explanation for the lack of correlation between circulating thyroid hormone and TSH concentrations in patients with CH (Fig. 38.1). It is well documented that glycosylation plays a fundamental role in modulating the expression of TSH bioactivity (7,14,15,16). Impaired control of TSH synthesis and secretion by TRH, and other neuroendocrine or paracrine factors, may be associated with alterations of posttranslational processing of the molecule, resulting
in the release of TSH forms with altered glycosylation and variable bioactivity (8,10,14,15,16). Indeed, the TSH levels measured in sera of CH patients are the likely result of the maximal secretory activity from the residual number of functional thyrotrope cells. In such conditions, the secretion of molecules with prolonged half-life, such as highly sialylated TSH, may represent an advantage for residual thyrotrope cells (8,15).
in the release of TSH forms with altered glycosylation and variable bioactivity (8,10,14,15,16). Indeed, the TSH levels measured in sera of CH patients are the likely result of the maximal secretory activity from the residual number of functional thyrotrope cells. In such conditions, the secretion of molecules with prolonged half-life, such as highly sialylated TSH, may represent an advantage for residual thyrotrope cells (8,15).
The relevant contribution of both reduced thyrotrope population and decreased biological activity of secreted TSH was confirmed by the biochemical and pathological characteristics of the animal model affected with pure hypothalamic hypothyroidism, the TRH knockout mice (17), which confirmed the relevant role played by TRH action in the regulation of the pituitary–thyroid axis (9,10,18). No TRH gene defect has been documented so far in humans, but defective TRH action due to natural mutations in the TRHR gene have been described in two families (19,20). Interestingly, the early development of patients with complete TRH resistance appeared uneventful and the diagnosis in the male proband with homozygous TRHR mutations was reached because of delayed growth at 11 years of age (20) (Table 38.2). Study of these families with complete resistance to TRH action demonstrated that the hypothalamic hormone is required to set the pituitary feedback mechanism at a level adequate to maintain thyroid hormone levels in the normal range. In addition, the conservation of a significant nocturnal TSH surge in this condition indicates that TRH action influences the amplitude, but is not required for the determination of the circadian oscillation.
TSHβ gene defects are a frequent cause of inheritable isolated CH (21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44). Missense, truncating, and intronic variants have been described (Fig. 38.2). Most of them cause a defective dimerization with the α-GSU, thus preventing the production of a complete and bioactive TSH molecule. As a result of this impaired dimerization, the finding of an excess of circulating α-GSU levels in a patient with isolated congenital CH is pathognomonic of a TSHβ gene defect (30). With the increasing reports of TSHβ mutations, several were found to cluster in a “hot spot” involving codon 105 in exon 3 in either Europe or South and North America, thus supporting the existence of a common ancestor in several of these cases. This variation results in a truncated TSHβ subunit lacking the so-called “seat-belt” domain, which is involved in the stabilization of the glycoprotein hormone heterodimer.
In patients with defects in the pituitary transcription factor genes (Table 38.2), CH is due to a defective differentiation of pituitary cells (45). Nevertheless, in several cases of mutations in the PROP1 or POU1F1 genes, CH may have a delayed onset during childhood suggesting that defects may also affect thyrotrope cell survival rather than commitment or differentiation. Defects of leptin signal due to leptin receptor mutations may in some cases cause a typical hypothalamic defect (46). Thus, leptin signaling was indeed shown to control TRH expression in hypothalamic neurons (47). These data and additional evidence in experimental models show that TRH neurons play a pivotal role in the pathways regulating metabolism and energy expenditure (47,48). The different combination of pituitary hormone defects and/or the association with particular complex phenotypes suggest the involvement of specific inheritable defects (Table 38.2) (45).
Pituitary tumors represent the most frequent cause of acquired CH, accounting for more than half the cases (49).
Among them, pituitary adenomas may be nonfunctioning or hypersecreting GH, prolactin (PRL), or, less frequently, ACTH or gonadotropins. Varying degrees of hypopituitarism may result from compression of normal pituitary cells. The pituitary stalk and the hypothalamus may be directly damaged by the suprasellar extension of the tumor. Metastases to the hypothalamic–pituitary region arising from carcinomas of the breast, lung, and occasionally other sites are infrequent and usually reflect the presence of advanced disease.
Among them, pituitary adenomas may be nonfunctioning or hypersecreting GH, prolactin (PRL), or, less frequently, ACTH or gonadotropins. Varying degrees of hypopituitarism may result from compression of normal pituitary cells. The pituitary stalk and the hypothalamus may be directly damaged by the suprasellar extension of the tumor. Metastases to the hypothalamic–pituitary region arising from carcinomas of the breast, lung, and occasionally other sites are infrequent and usually reflect the presence of advanced disease.
Extrasellar brain tumors can also cause CH by affecting hypothalamic function (49,50). Hypopituitarism is usually accompanied by diabetes insipidus. Craniopharyngioma is the most frequent extrasellar brain tumor causing hypopituitarism and CH, especially in younger patients. Meningiomas, gliomas, Rathke’s cleft cyst, empty sella, and non-tumorous mass lesions are less frequent CH causes.
Medical interventions for the treatment of sellar and extrasellar tumor masses represent an additional risk for CH. External radiotherapy for tumors of the head and neck have been described which affect the hypothalamus, the pituitary, and the thyroid, and hypothyroidism often results from damage of one or more of these structures (the so-called “mixed hypothyroidism”) (50,51,52,53,54,55). Hypothyroidism resulting from pituitary or hypothalamic dysfunction had been reported in 20% to 50% of patients irradiated for nasopharyngeal or paranasal sinus tumors (56) and in about 65% of patients irradiated for brain tumors (51,52). Overall, the risk of the development of central hypothyroidism is related to the total radiation dose, being higher for intracranial solid tumors, which are treated with higher radiation doses (52,55). The technical improvements in radiotherapy instrumentation and earlier diagnosis are expected to reduce the development of CH in a number of cancer survivor patients (52). TSH deficiency can also result from direct irradiation of the pituitary, by either conventional external radiotherapy or γ-knife for pituitary tumors. If not present initially, CH may result from surgical therapy of pituitary tumors. The dimension and extrasellar extension of the pituitary mass, as well as the neurosurgeon’s expertise are critical factors in this context.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


