 Figure 37.1 Esthesioneuroblastoma in a 9-year-old girl presenting with nasal obstruction and pain. MRI axial (A) and coronal (B) STIR (short tau inversion recovery) images show a mass in the left sinonasal cavity, involving the anterior left ethmoid air cells, the anteromedial aspect of the left maxillary sinus and the left nasal cavity. Sagittal (C) and coronal (D) CT images show thinning of the cribriform plate (white arrow) and the presence of calcifications (black arrow). |
TABLE 37.1 Esthesioneuroblastoma—Hyams Histopathologic Grading System | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
TABLE 37.2 Esthesioneuroblastoma—Modified Kadish Staging System | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
with oropharyngeal cancer regardless of the use of tobacco or alcohol. The incidence of HPV-associated oropharyngeal cancer has significantly increased since 1973, particularly among white males aged 40 to 59 years.31 Approximately 20% to 25% cases of head and neck cancer contain oncogenic HPV, mostly types 16, 31, and 33. In a study of 1,235 children, the prevalence of HPV in the oral cavity was 1.9% and the highest rates were seen in patients younger than 1 year and in those aged between 16 and 20 years.32 HPV-associated oral cancers tend to occur in younger patients of high socioeconomic status, are associated with sexual behavior, more often involve the lingual and palatine tonsils, frequently have poorly differentiated basaloid features, express p16, and have a better prognosis and response to radiotherapy than other head and neck carcinomas.33,34,35 Current preventive strategies that incorporate vaccine programs targeting the adolescent population may prove beneficial in decreasing the incidence of cervical and head and neck HPV-associated cancers.36,37
 Figure 37.2 Pleomorphic adenoma of the right parotid gland in a 13-year-old male. Axial (A) and coronal (B) CT images disclose a well-circumscribed right retropharyngeal mass (white arrows) with calcifications (black arrow). |
million per year.57 Although representing approximately 1% of all pediatric malignancies, it accounts for 35% to 50% of all nasopharyngeal malignancies. NPC originates from the surface epithelium and differs from other head and neck carcinomas by its very distinct histologic, epidemiologic, and biologic characteristics. NPC has an endemic distribution among well-defined ethnic groups, such as inhabitants of some areas of Southeast Asia and in Alaskan Eskimos, where the incidence is 25 to 50 and 15 to 20/100,000 persons per year, respectively.58
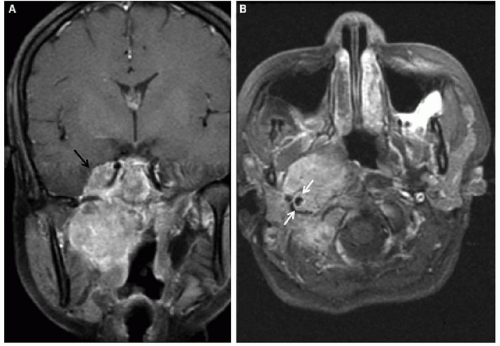 Figure 37.3 NPC in a 16-year-old Asian American female presenting with nasal obstruction, headache, and cervical lymphadenopathy. Coronal (A) and axial (B) T1-weighted images demonstrate a large right nasopharyngeal mass obliterating the right parapharyngeal space. The mass extends posteriorly to the carotid sheath (white arrows), inferiorly to the level of the oropharynx, and superiorly to the cavernous sinus and right middle cranial fossa (black arrow). |
cells containing the virus. Staging of NPC usually follows the TNM system, which has been shown to be predictive of outcome, and very helpful in defining therapy (Table 37.3).64
TABLE 37.3 AJCC Staging System for Nasopharyngeal Carcinoma | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
durable responses, and patients with EBV+ recurrent NPC should be considered for this type of therapy in combination with a taxane-containing salvage regimen.69
TABLE 37.4 Treatment of Advanced Childhood Nasopharyngeal Carcinoma | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||
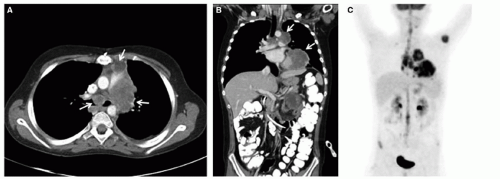 Figure 37.4 NUT-midline carcinoma in a 10-year-old boy. Axial (A) and coronal (B) CT images show a large left para-aortic and para-cardiac mass (arrows) with invasion of the left inferior pulmonary vein and lateral wall of the left ventricle, and extending into the anterior mediastinum and subcarinal space. PET scan (C) demonstrates intense FDG avidity and the presence of a left humeral metastasis. |
mass, malocclusion, paresthesias, pain, and evidence of unerupted teeth in the affected area.73,75 Analysis of small numbers of tumors suggest that p53, MDM2, and p14 may be responsible for tissue structuring and cytodifferentiation in ameloblastoma.74 In addition, recent reports have documented the existence of a complex interaction that favors tumor formation and is mediated by the secretion of frizzled-related peptide (sFRP2), which impairs bone formation, as well as IL-6 and RANKL, which promote bone resorption.76,77
recurrent pneumonia, or hemoptysis and mediastinal and distant metastases are often present at diagnosis.113,116 Treatment recommendations for children with lung carcinomas should be made in consultation with adult oncologists who are experienced in treating these histologies.
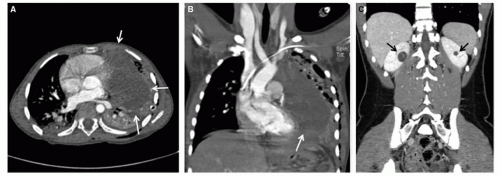 Figure 37.5 DICER1 syndrome. A and B: Six-year-old girl presenting with chest pain and respiratory distress. Family history was significant for thyroid nodules in the father, who underwent thyroidectomy during adolescence. Axial (A) and coronal (B) CT images demonstrate a large left Type III pleuropulmonary blastoma (white arrows). C: Sixteen-year-old female with history of vaginal/cervical rhabdomyosarcoma at 4 years of age. Coronal CT shows bilateral hypoattenuating cystic masses with enhancing septations (black arrows). Partial nephrectomies revealed cystic nephroma. Both patients tested positive for germ-line DICER1 mutations.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|