diagnosis, the ultimate reference point for all diagnostic procedures, and continuing through immunohistochemistry (IHC), cytogenetics, fluorescence in situ hybridization (FISH), single nucleotide polymorphism (SNP) arrays, polymerase chain reaction (PCR), and whole genome methods, will be discussed in turn. The final portion of the chapter discusses the future implications of the increased availability of novel, often targeted, therapeutics and diagnostic methods on the necessary approach to pediatric cancer diagnosis. It should come as no surprise that diagnostic pathology in the 21st century will bear little resemblance to that in the 20th century.
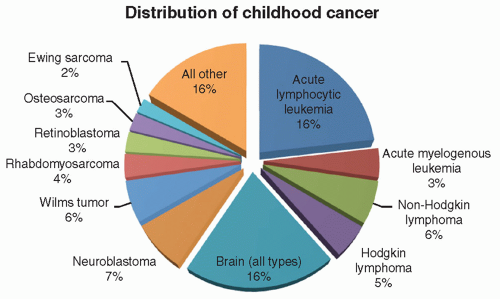 Figure 8.1 Distribution of childhood cancer. The first four slices of this pie chart account for approximately one-third of childhood malignancy and are accounted for by acute lymphocytic leukemia (approximately one-fourth) and acute myelogenous leukemia, non-Hodgkin lymphoma, and Hodgkin disease (the remaining three wedges). The next most common category is brain tumors, which account for approximately 20% of all childhood tumors. This is followed by neuroblastoma and Wilms tumor, which are roughly equal in incidence. Rhabdomyosarcoma, retinoblastoma, osteosarcoma, and Ewing sarcoma account for the remaining four defined wedges of the pie chart. All other tumors account for less than 20% of all childhood tumors in aggregate. Notably absent from the previous is any significant incidence of carcinoma, the most common form of adult cancer. |
have documented other less-striking differences in tumor-type incidences among ethnic populations.4,32 Knowledge of a patient’s ethnicity can be a helpful adjunct in diagnosis, particularly with undifferentiated childhood tumors, such as an African American child with a differential diagnosis of NB versus EFT. Such a patient is far more likely to have NB, based on these criteria.
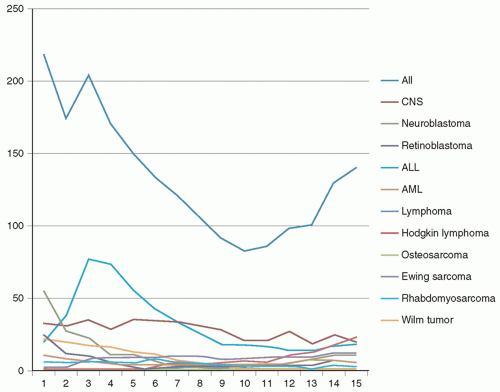 Figure 8.2 Childhood cancer: age incidence. That childhood cancer is largely embryonal in character is immediately evident from this chart of age incidence. The top line, which represents all forms of childhood cancer, shows a nearly linear increase from birth until approximately 10 years of age at which time the slope of the line reverses and begins to climb steadily toward adult-type incidences found later in life, largely due to the appearance of adult-type malignancies (not detailed here). The overall high incidence at birth is largely attributable to neuroblastoma, which generally presents in the first year or two of life. The large peak seen at approximately 3 years of age is due to the peak incidence of the most common form of childhood cancer, acute lymphocytic leukemia. Because of the lesser frequency, all the other tumors seem to be relatively constant in incidence (e.g., brain tumors, the next most common malignancy seen here), but in reality, striking differences in ages are found here as well. |
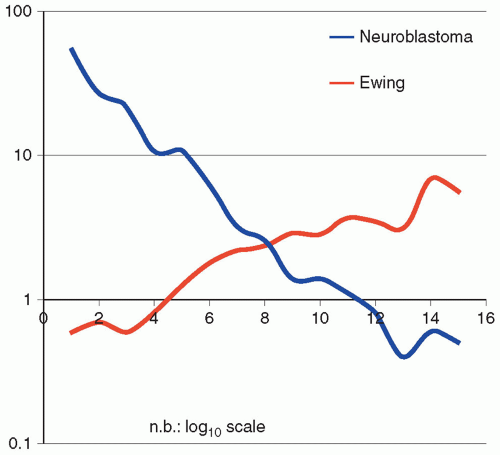 Figure 8.3 Neuroblastoma versus Ewing sarcoma: age-specific incidence. Although the two most common neural tumors in childhood are neuroblastoma and Ewing sarcoma, respectively, they are strikingly different in their incidence and character (age, incidence rate), despite their common phenotype. This is evident from this graph in which neuroblastoma is clearly a largely congenital tumor most common at birth and declining to extreme rarity by the age of 7, versus Ewing sarcoma with a nearly linear increase from birth when it is exceedingly uncommon to the age of 14 when it reaches a maximum in the pediatric age group. From this, one would assume that the genetic origins for the two phenotypically similar tumors are distinctly different. Note that the incidence data are plotted on a log10 scale. |
ancillary diagnostic procedures performed to detect gene translocations, antigen expression, or gene amplification, respectively. The availability of advanced diagnostic procedures common to childhood cancer evaluation is unusual in adult cancer centers, a striking difference often overlooked by adult cancer diagnosticians. This is unfortunate because these advanced diagnostic procedures require special handling (e.g., fresh viable tumor tissue for short-term culture and cytogenetics, fresh frozen tissue for flow cytometry and molecular analysis) that are generally not part of the routine diagnostic workup for adult cancers. In many cases, local institutions lack the facilities to perform these advanced diagnostic studies. In this situation, tissue can still be handled according to prescribed protocols that ensure proper tissue handling and transportation to a central reference laboratory that performs the appropriate studies. The COG, which treats the majority of children with cancer in North America, maintains a central reference facility, the Biopathology Center, in Columbus, Ohio, which receives these specimens from the nearly 250 participating institutions, and offers a number of these advanced diagnostic procedures, as well as central pathology review for many of the treatment protocols. If the primary pathologist is not familiar with the proper protocol or diagnostic procedures required, the opportunity to provide additional diagnostic, treatment, and prognostic information derived from specialized tests may be lost. It is useful to have a well-defined protocol and action plan for childhood tumor diagnosis to avoid loss of critically needed diagnostic information.
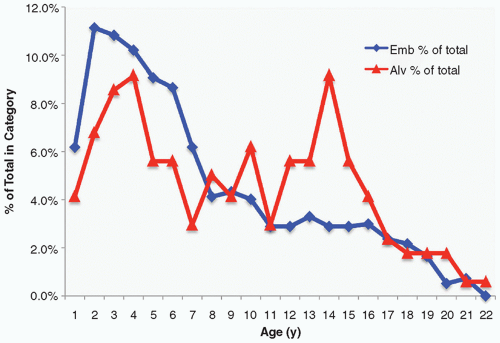 Figure 8.4 Rhabdomyosarcoma: age-specific incidence. Another childhood neoplasm with a striking variation in incidence by age is childhood alveolar rhabdomyosarcoma, which although less common than embryonal rhabdomyosarcoma is nonetheless an important source of fatality in this disease because of its worse prognosis. When plotted as percentage of total cases, alveolar rhabdomyosarcoma shows a bimodal age incidence, the first mode roughly corresponding to the peak age incidence of embryonal rhabdomyosarcoma, and the second mode with no parallel increase in embryonal rhabdomyosarcoma seen at approximately 14 years of age. Studies have suggested that the two modes are in fact genetically distinct as well; the first mode includes mostly PAX7-FOXO1A tumors, while the second is largely PAX3 (see text). |
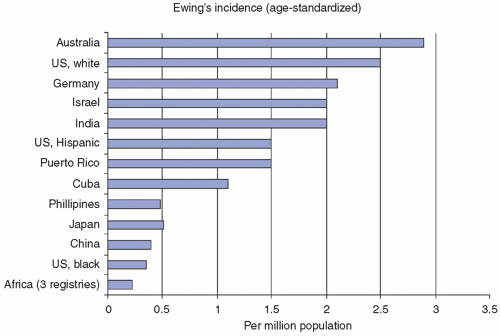 Figure 8.5 Ewing sarcoma family of tumors: ethnicity versus incidence. U.S. SEER data from the National Cancer Institute documents striking differences in the incidence of Ewing sarcoma family of tumors among different ethnic groups. Australia reports the highest incidence on a per-million-population basis, whereas three different African registries report the lowest incidence reported anywhere in the world. U.S. blacks and other Asian populations also have an extremely low incidence of Ewing sarcoma. These observations have strongly suggested a genetic component to Ewing susceptibility, but the genetic predisposing factors have not yet been identified. (Data courtesy of Jonathan Buckley, MD, PhD, Preventive Medicine, Keck School of Medicine, USC.) |
has limited utility for many molecular genetic diagnostic procedures and may limit conventional diagnostic techniques, such as immunocytochemistry and PCR. In an attempt to promote nonroutine tissue handling, many childhood cancer protocols specifically request submission of fresh frozen or even viable tumor tissue. Despite these efforts, no protocol ever achieves perfect compliance with these protocol requirements. As a result, many of the necessary diagnostic procedures have been (and continue to be) adapted to formalin-fixed paraffin-embedded (FFPE) tissue. FISH on FFPE tissue for MYCN amplification instead of frozen tissue for DNA extraction and Southern blotting in NB is one example. More recently, so-called whole-genome and next-generation sequencing methods (discussed later in this chapter) have been adapted for use with FFPE tissue.41,42,43,44,45,46,47 The simple fact that the majority of tumor tissue is formalin fixed and embedded in paraffin blocks has forced the development of these methods, to great advantage for pathologists, who otherwise would be precluded from using molecular methods on the majority of cases.
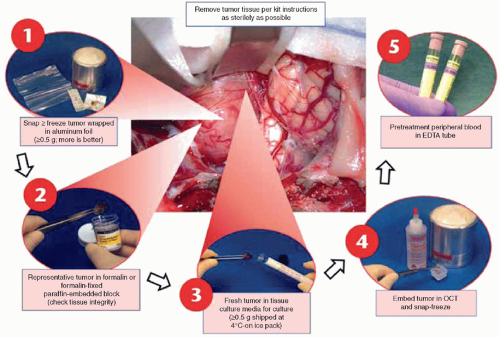 Figure 8.6 Tissue handling diagram. Tissue for optimal pathologic and biologic studies requires specialized handling as summarized in this chart. This diagram, which has been used by participating institutions in the Children’s Oncology Group for over 15 years, has been widely adopted and led to significant improvements in diagnostic accuracy in childhood cancer. Note that fresh tissue, not fixed, is required at the outset, for steps 1, 3, and 4. A pretreatment peripheral blood (for DNA from buffy coat and protein from plasma) is required for step 5, and will allow comparison of tumor versus normal DNA, an increasingly important part of the cancer diagnostic workup (see Molecular Diagnostics section). (Diagram created by Steve Qualman, MD, for Children’s Oncology Group.) |
TABLE 8.1 Top 10 Diagnostic Methods for Tumor Diagnosis | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
oncogene, a member of MYC family genes, is tested by FISH and found to be amplified in approximately 20% of NB cases (Fig. 8.8) Amplified MYCN is transcribed and translated, and expressed as an excessive amount of MYCN protein. MYCN protein then forms a heterodimer with MAX (MYC-associated factor X) protein and activates various downstream genes related to cell proliferation, differentiation inhibition, and apoptosis, etc., and results in aggressive clinical behavior.60,61 Recently, MYC (aka C-myc) protein expression has been detected immunohistochemically in some of the undifferentiated NB tumors, especially when the MYCN gene is not amplified. Both MYCN and MYC protein-expressing tumors are often associated with prominent nucleolar formation,62 the putative site of RNA synthesis and accumulation (Fig. 8.9), and patients with those MYC (both MYCN and MYC)
protein-driven NB tumors seem to have a very poor clinical outcome.63 DNA ploidy, 1p LOH, and 11q LOH are also important for diagnosis, treatment, and prognosis.64 SNP arrays have been employed to detect prognostic DNA copy number variation on chromosomes 1, 10, and 17.65,66,67 Each of these factors plays an integral part in determining whether a child with NB will be placed on a high-risk, intermediate-risk, or low-risk protocol. More recently, ALK gene mutations/overexpression, ATRX gene mutation, and LIN28B mutation/aberration have been noted to affect prognosis, independently of other factors.68,69,70 Most recently, a series of reports focusing on NB cells and their microenvironment have been published, which noted that, like other neoplastic diseases, cross-talk between tumor cells and nontumor cells, such as tumor-associated macrophages (TAMs), plays a significant role in promoting metastatic progression of NB tumors.71,72
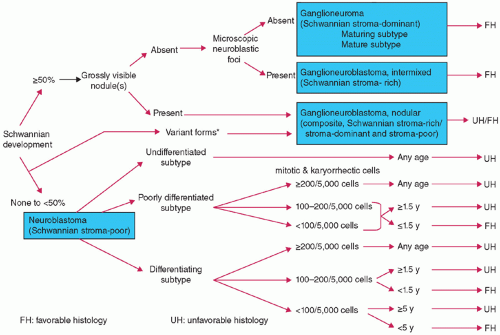 Figure 8.7 International Classification System for Neuroblastoma Prognosis and Diagnosis. Flowchart for prognostic evaluation: There are two groups for the final evaluation of pNTs: they are favorable histology (FH) and unfavorable histology (UH). Prognostic evaluation according to the International Neuroblastoma Pathology Classification is applicable to only those tumors (either primary or metastasis) that are biopsied or surgically excised prior to chemotherapy/irradiation therapy. Those cases with difficulty in making prognostic evaluation can be grouped into “Not Evaluable” category with some note such as “due to a limited amount, limited quality, etc.” Surgical pathology report for the cases whose tumor tissue is obtained after chemotherapy/irradiation therapy should have a clear statement of “Status Posttreatment” in their diagnosis line. (Courtesy: Dr. Hiroyuki Shimada, Children’s Hospital Los Angeles.) |
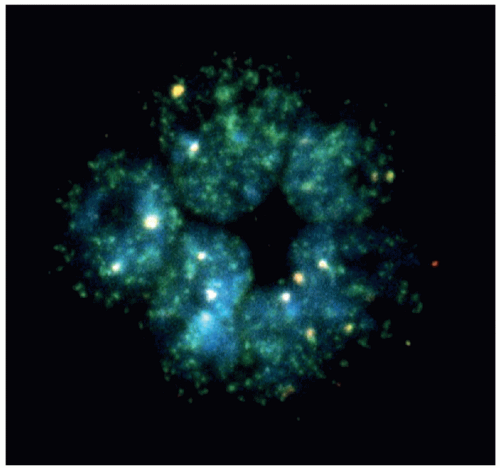 Figure 8.8 MYCN amplification in neuroblastoma detected by fluorescence in situ hybridization. A touch preparation of a neuroblastoma was incubated with a fluorescent probe for MYCN. Here one can readily see hundreds of signals in a tumor rosette, each representing at least one copy of MYCN. This clearly documents MYCN amplification in these tumor cells, denoting a poor prognosis group (MYCN amplified). |
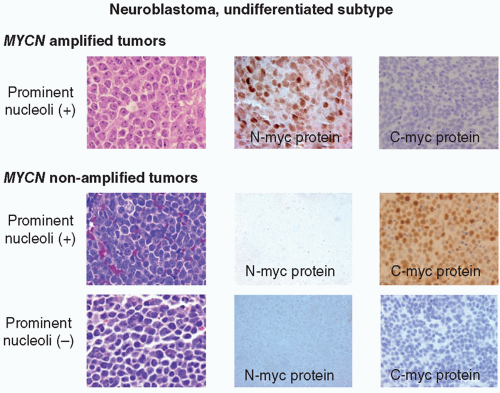 Figure 8.9 Neuroblastoma morphology and MYC protein expression. MYCN-amplified undifferentiated NB tumors often have prominent nucleolar formation and N-myc protein expression. Among MYCN nonamplified undifferentiated NB tumors, those with prominent nucleoli tend to express C-myc protein, and others with typical salt-and-pepper nuclei are negative for both N-myc and C-myc protein expression. (Figure kindly supplied by Dr. Hiroyuki Shimada, op cit.) |
TABLE 8.2 Differential Diagnosis of Pediatric Bone and Soft Tissue Solid Tumors | |
|---|---|
|
group, they have a primitive or embryonal appearance, often present in misleading clinical locations like bone marrow metastases from an occult primary, and lack specific morphologic features that allow for a precise diagnosis without ancillary methods.73,74
 Figure 8.10 Histopathology of typical small round cell tumors. Historically, Ewing sarcoma (A), rhabdomyosarcoma (B), neuroblastoma (C), and lymphoma (D) were difficult to distinguish from one another, particularly when undifferentiated rhabdomyosarcoma or neuroblastoma was encountered, as illustrated here. None of the four tumors show light microscopically discernible evidence of differentiation and therefore histogenesis, which is critical to a diagnosis by light microscopy. |
a differential diagnosis is formulated and a series of special studies are selected to provide a definitive diagnosis. The most commonly used ancillary diagnostic method is immunocytochemistry. Either concurrent with or subsequent to immunocytochemical study, special histochemical stains (periodic acid-Schiff [PAS], reticulin, trichrome), EM, and molecular studies may be initiated. Findings from all of these studies are reviewed and interpreted in concert with the clinical history and diagnostic imaging findings. With the vast majority of SRCTs, this multimodal approach will yield a precise and definitive diagnosis that directs further surgical intervention, oncologic management, follow-up, and prognosis. Using these extensive and coordinated methods, it is rare that diagnoses are modified by additional testing, consultative review by pathologists at other institutions, or by expert pathologist reviewers for COG protocol studies.
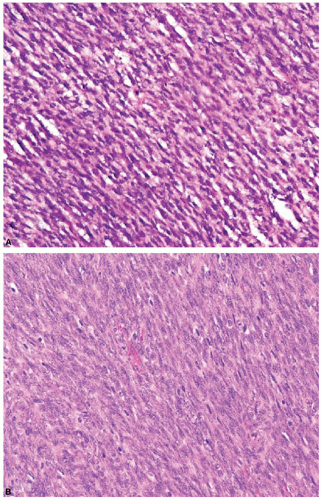 Figure 8.11 Poor (A) and well-done (B) histology of a typical soft tissue tumor. The first panel (A) illustrates the appearance of a soft tissue neoplasm in a 9-month-old infant. The first material is from the frozen section; the second, after formalin fixation and paraffin embedding. The correct interpretation only became apparent with proper fixation and embedding as seen in Panel B, in which the correct identity as congenital fibrosarcoma became evident. Many diagnostic problems in childhood tumor interpretation benefit from optimal quality histology and can be the useful and necessary first step in the choice of other ancillary diagnostics to confirm the impression gleaned from careful scrutiny of a well-prepared hematoxylin and eosin section. (Photomicrographs kindly provided by Dr. Larry Wang, Children’s Hospital, Los Angeles.) |
An antigen-retrieval solution (10 mM citrate buffer at pH 6.0; 100 mM Tris-HCl at pH 8.0; 1 mM EDTA-NaOH at pH 8.0; 5% urea) and heat treatment (microwave, microwave and pressure cooker, autoclave, pressure cooker, water bath, steamer, thermal cycler) allow reversing protein cross-linking by formalin fixation. Optimal antigen-retrieval intensity from FFPE tissue has been achieved with heat treatment at temperatures of 10°C for 20 minutes, 90°C for 30 minutes, 80°C for 50 minutes, and 70°C for 10 hours. After antigen retrieval, enzyme digestion (trypsin, pronase, proteinase K, pepsin) is usually not necessary prior to reaction of the tissue with the antibody but may be needed for certain antibodies. For most antibodies, heat treatment for 20 to 30 minutes at 100°C with either citrate buffer at pH 6.0 or Tris-HCl buffer at pH 7.0 to pH 8.0 are effective for antigen retrieval. The antibody manufacturer provides general guidelines; however, it is necessary to determine the optimal conditions for the antibody in each institution’s immunocytochemistry laboratory. In addition, monitoring for accurate immunoreactivity requires that known control tissue be run parallel, or on the same glass slide as the patient’s tissue, and under the same antigen retrieval conditions. Immunoreactivity may be adversely affected if the tissue sections are prepared sometime in advance of staining. Tissue sections should be prepared and undergo processing for immunostaining without delay to avoid antigen degradation. It is also possible to preserve antigenicity if the tissue sections on glass slides are stored at -70°C until immunocytochemistry is performed.
 Figure 8.12 Immunocytochemistry by cellular localization. Immunocytochemistry is widely utilized on a daily basis in surgical pathology of adult and childhood tissues. In the case of childhood cancer, many specific classes of antigens are detectable. Illustrated here are three categorical examples: (A) an example of nuclear staining for a transcription factor, MyoD, widely used to detect extremely undifferentiated childhood rhabdomyosarcoma; (B) vimentin, ubiquitous intermediate filament found in all mesenchymal tissues and their tumors counterpart. The staining is localized to the cytoplasm. This antigen is most used to verify antigenic preservation sample and is not specific for any given tumor; (C) surface antigen detection, MIC2 (CD99) in Ewing sarcoma. Here apparent cytoplasmic as well as cell surface localization is detectable. Such antigens routinely show surface staining that is difficult to photograph but easily detected by focusing up and down through the sample under the light microscope. Many of these antigens are also quite labile and not readily detected in routinely processed tissue. |
TABLE 8.3 Immunocytochemical Antibodies of Diagnostic Importance in Childhood Tumors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
1. Staining at tissue section edges (trapping) or in crevices, while the tissue away from the periphery and tissue section defects has no significant immunoreactivity.
2. Focal staining due to trapping of reagent in an elevated or depressed region of the tissue. It is possible that the tumor is composed of variable cell types with different immunoreactivity to the antibody, resulting in an island of tumor cells with focal staining; however, the staining pattern should follow the cytoplasmic, nuclear, or membranous pattern characteristic for the particular antibody interaction with a specific tumor type. Aberrant expression of a particular antigen(s) by tumor cells may occur (Table 8.5).
3. Diffusion or “leaking” of antigenic proteins from surrounding normal tissue that has been infiltrated by the SRC T. In particular, false-positive immunoreactivity with myogenic antibodies has been reported when an SRC T has invaded skeletal muscle.
4. Poor techniques, such as incomplete paraffin removal, high-melting-point paraffin, prolonged or excessive heat with antigen retrieval, poorly fixed or necrotic tissue, thick section preparation, endogenous biotin with inadequate peroxidase/biotin blocking, incomplete rinsing of slides, desiccation of tissue sections during processing, inadequate incubation time, chromogen staining too intense, and inappropriate concentration of antibody are all factors that may lead to spurious and misleading immunocytochemical findings and interpretations.
TABLE 8.4 Immunocytochemistry Approach to Small Round Cell Tumors of Childhood | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
this feature is determined by immunocytochemical and ultrastructural investigations (Tables 8.4 and 8.5). Typically, this tumor expresses desmin in a “dot-like,” “globoid” or Golgi-like cytoplasmic pattern, NSE, and cytokeratin. Several other neural, myogenic, and epithelial antigens may also be expressed. Of particular interest is the immunoreactivity with tumor suppressor antibody WT-1. The Ewing sarcoma marker, CD99, may also be expressed in a limited number of cases. In the absence of desmin and cytokeratin staining, the misdiagnosis of extraosseous Ewing sarcoma could be rendered. Typically, small cell (undifferentiated) osteosarcoma (Tables 8.4 and 8.5) reacts with only two readily available antibodies, vimentin and retinoblastoma protein, and it may react with
p53. Aberrant immunoreaction with smooth muscle actin, Leu7 (CD57), and S-100 protein may confuse this neoplasm with other poorly differentiated peripheral nerve sheath tumors or mesenchymal sarcomas. The small cell variant of hepatoblastoma (Tables 8.4 and 8.5) may be confused with metastatic NB or primary hepatic NB in stage IV-S disease. This tumor has several overlapping features with NB in that both may immunoreact with NSE and chromogranin. In contrast, small cell hepatoblastoma should express cytokeratin and epithelial membrane antigen, and may also immunoreact with alpha-fetoprotein, beta-human chorionic gonadotropin (HCG), carcinoembryonic antigen (CEA), and alpha-1-antitrypsin. Interestingly, small cell hepatoblastoma has been shown to lack nuclear INI1/SMACRB1 expression, similar to rhabdoid tumor.111 Blastemal predominant WT (Tables 8.4 and 8.5) may be somewhat difficult to diagnose if the presence of a kidney tumor is not known, and metastatic disease at another site, such as the lung, is considered to be a primary lesion by the clinician. This tumor may have a very undifferentiated appearance and may mimic any of the SRCTs morphologically. Immunocytochemical studies of blastemal WT should demonstrate pancytokeratin and vimentin expression. In addition, more than 40% of WTs will immunoreact with WT-1 antibodies. MPNST (Tables 8.4 and 8.5) may also resemble SRCTs; however, it will typically express markers associated with peripheral nerve derivation, such as S-100 protein and Leu7. It may also immunoreact with HMB-45 and epithelial antibodies. Aberrant immunoreactivity to desmin, muscle-specific actin, and CD68 may confuse this tumor with other SRCTs. It is also well known that rhabdomyoblastic cells may be seen in MPNSTs (triton tumors), and this tumor could be confused with a poorly differentiated spindle cell RMS or malignant ectomesenchymoma. Synovial sarcoma (Tables 8.4 and 8.5) tends to express the epithelial markers, cytokeratin and epithelial membrane antigen, and the intermediate filament, vimentin. In addition, bcl2 and CD99 may also be identified. Confusion with peripheral nerve sheath tumors may occur when SS immunoreacts with S-100 protein. An aberrant expression of CD99 may be seen in this tumor. In poorly differentiated SS, there may be unexpected expression of Leu7 (CD57), nerve growth factor receptor, CD56, type IV collagen, and neurofilament triple protein. There may be a gray zone between poorly differentiated SS and MPNST that can only be resolved definitively by molecular studies for the translocation characteristic for SS [t(X:18), SYT/SSX]. Immunocytocytochemical expression of TLE1 in SS has been proposed as a unique marker of SS, but this has been recently questioned.112 Extrarenal rhabdoid tumor (Tables 8.4 and 8.5) may mimic RMS; however, this tumor typically expresses cytokeratin and vimentin in a particular pattern, and lacks nuclear expression of IN1/SMARCB1.113 The cytoplasm is engorged with intermediate filaments that displace the nucleus toward the periphery. Rhabdoid tumors may express desmin and muscle-specific actin, whereas RMS may aberrantly immunoreact with pancytokeratin and epithelial membrane antigen. The characteristic intermediate filament pattern by immunocytochemistry and EM is useful in differentiating these tumors from one another.
TABLE 8.5 Immunocytochemical Workup for Undifferentiated Childhood Tumor | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
 Figure 8.13 Tissue microarray (TMA). This TMA with more than 100 cores of childhood rhabdomyosarcoma (RMS) and related control tissues has been reacted with an antibody (TFAP2β) that reacts only with the fusion-positive (e.g., PAX-FOXO1A, PAX 3 or 7) alveolar rhabdomyosarcoma (ARMS). The ARMS fusion-positive cores are highly reactive; embryonal RMS and fusion-negative ARMS are not, as is also true of other tumors on the array and normal control tissues such as skeletal muscle. Thus, with one slide, an antibody can be evaluated for its specificity and sensitivity, as shown here. (Courtesy of Mike Anderson, Children’s Hospital, Los Angeles, Keck School of Medicine, USC.) |
for maintaining cytoplasmic glycogen are not currently a standard of practice. The utility of the PAS stain in tumor diagnosis has lost much of its sensitivity and specificity in the face of far more tumor-specific diagnostic methods like IHC and molecular genetic methods. Still, a strongly positive PAS stain in a suspected Ewing tumor versus lymphoma versus neuroblastoma is still strong evidence for a diagnosis of Ewing tumor. When EM is not available, CD99 results are equivocal, molecular methods yield conflicting or no result, and histopathology is equivocal, a strong positive PAS stain can lend credence to a diagnosis of Ewing sarcoma over the alternatives.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


