TABLE 32.1 Distribution of Histologic Subtypes of Soft Tissue Sarcoma Derived from the Surveillance, Epidemiology, and End Results Database in Children and Adults in the United States, 1993 to 2002 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Werner syndrome. Patients with neurofibromatosis type 1 (NF-1) have a significantly increased risk of developing MPNST, with a lifetime risk estimated to be in the 6% to 13% range.7 Leiomyosarcoma occurs at greater than background levels in children with acquired immunodeficiency syndrome (AIDS); Epstein-Barr viral infection is thought to play a causal role in the development of this cancer. Desmoid fibromatosis has been reported to develop in about 10% of patients with familial adenomatous polyposis (FAP); the risk is greater for those with APC gene mutations that occur 3′ of codon 1444 and for those who undergo abdominal surgery.8
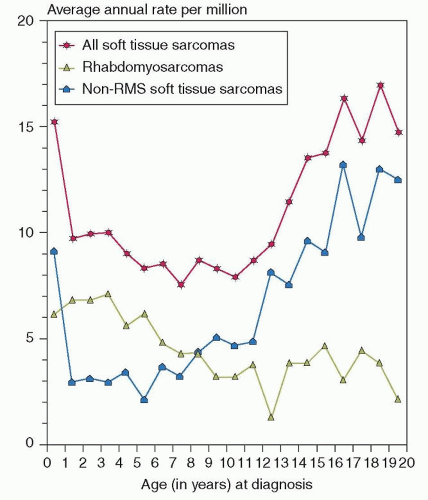 Figure 32.1 Graph shows the annual incidence of STSs by year of age using data from 1976 to 1994. RMS is more common from 1 to 8 years of age, whereas NRSTS is more common in infants younger than 1 year and in children older than 8 years. (From Gurney JG, Young JL, Roffers SD, et al. Soft tissue sarcomas. In: Ries LAG, Smith MA, Gurney JG, et al., eds. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program. NIH Pub. No. 99-4649. Bethesda, MD, 1999.) |
TABLE 32.2 Genetic Aberrations in Childhood Nonrhabdomyosarcoma Soft Tissue Sarcomas | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Extent of disease (nonmetastatic vs. metastatic)
Histologic grade (low vs. high)
Size of the primary tumor (≤5 cm vs. >5 cm)
Extent of surgical resection (resected vs. unresected).
those with large, high-grade tumors tend to develop fatal distant disease dissemination. Patients in the low-risk category include those with nonmetastatic, resectable tumors that are either high grade and less than 5 cm in maximal diameter or low grade (any size). These patients have a long-term survival estimate of approximately 90%.
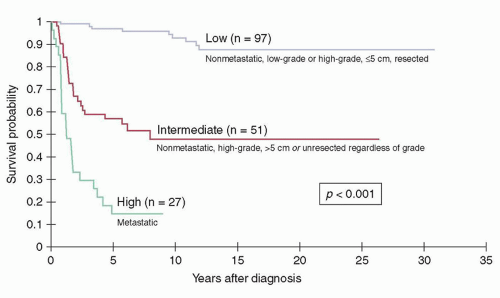 Figure 32.2 Kaplan-Meier survival distributions of patients treated at St. Jude Children’s Research Hospital according to risk group. (Figure constructed using data from Spunt SL, Poquette CA, Hurt YS, et al. Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: an analysis of 121 patients treated at St. Jude Children’s Research Hospital. J Clin Oncol 1999;17:3697-3705; Spunt SL, Hill DA, Motosue AM, et al. Clinical features and outcome of children with unresected non-rhabdomyosarcoma soft tissue sarcoma [NRSTS]. J Clin Oncol 2002;20:3225; and Pappo AS, Rao BN, Jenkins JJ, et al. Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children’s Research Hospital experience. Med Pediatr Oncol 1999;33:76.) |
children and adults, in some cases leading to differences in treatment recommendations. For example, fewer than 15% of pediatric GISTs harbor KIT or PDGFA gene mutations, whereas these mutations occur in about 90% of adult GIST. Based on these findings, upfront treatment for most pediatric patients is surgery alone, whereas most adults receive tyrosine kinase inhibitor therapy.31
draining the tumor. These lymph nodes are excised and then thinly sectioned for review by the pathologist.
resectability, most of these tumors will be of high grade, large, and invasive that would likely require RT based on pretreatment evaluation. The intent of preoperative RT is to shrink tumors and facilitate limb- or organ-sparing resection. The advantage of preoperative RT over postoperative RT is that the total dose delivered may be lower by 10% to 15% and the volume irradiated smaller, potentially resulting in fewer long-term effects on normal tissues. Another theoretical benefit to preoperative RT is that much of the irradiated tissue is resected, therefore potentially further reducing late effects of ionizing irradiation, particularly second cancers. Local treatment failure is a major challenge for patients with initially unresectable as compared with resected tumors.25 Spunt et al. showed a 44% cumulative incidence of local failure in 27 patients treated before 1987 where lower radiation doses (39.6 Gy) were thought to be a contributing factor; higher radiation doses (59.4 Gy) resulted in lower local recurrence rates (20%). Ferrari et al. reported the pooled results of over 300 patients with unresected pediatric NRSTS from United States and Europe research groups, and concluded that local progression or recurrence was the major cause of treatment failure, with RT correlating with a better outcome.46
radiation beam to allow direct high-dose, single-fraction RT to be delivered to the tumor bed with minimal toxicity.49 All of these contemporary technologies contribute to keeping the radiation therapy dose as low as possible, hence minimizing normal tissue exposure.
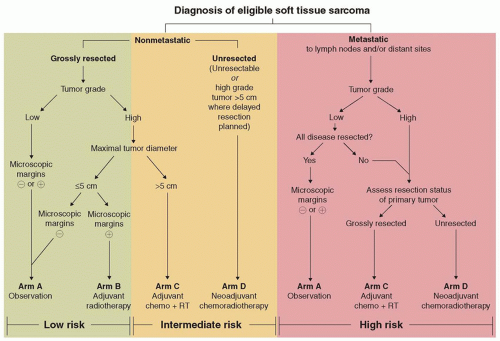 Figure 32.3 Risk group and treatment assignment algorithm for Children’s Oncology Group protocol ARST 0332, “Risk-Based Treatment for Non-Rhabdomyosarcoma Soft Tissue Sarcomas (NRSTS) in Patients Under 30 Years of Age.” (Used with permission, James Anderson, Children’s Oncology Group.) |
dose of ifosfamide in terms of disease-free or overall survival.56 Finally, the combination of gemcitabine and docetaxel has produced modest rates of response.59
bands.11 Delicate regularly distributed vessels with perivascular edema are distinctive. The tumor cells express vimentin and variable muscle-specific and smooth muscle actin, and up to 75% display nuclear beta-catenin reactivity.
TABLE 32.3 Clinical Features, Treatment Approach, and Outcome of Intermediate and Malignant Childhood Soft Tissue Sarcomas | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


