The rare inherited coagulation disorders (RICDs), usually transmitted in an autosomal recessive fashion, represent 3% to 5% of all the inherited deficiencies of coagulation factors and include fibrinogen, factor (F)II, FV, FV+FVIII, FVII, FX, FXI, FXIII, and the multiple deficiency of vitamin K-dependent factors.
1,2 Their distribution is variable in the world, with a prevalence in the general population of the presumably homozygous forms ranging from approximately 1 in 2 million for prothrombin (FII) to 1 in 500,000 for FVII deficiency (the most common) (
Table 53.1).
3 In countries with large Jewish communities, FXI deficiency is more prevalent. In Middle Eastern countries and Southern India, where consanguineous marriages are customary, autosomal recessive traits occur more frequently in the homozygous state.
4Data on the distribution of patients affected by RICDs are limited. Given their rarity, few centers have the possibility to follow and manage a consistent number of patients, so that scientific reports are usually limited to small numbers. Two recent large registries help to derive the worldwide distribution of RICDs: one by the World Federation of Haemophilia (WFH, www.wfh. org) and the other by the International Rare Bleeding Disorders Database (RBDD, www.rbdd.org).
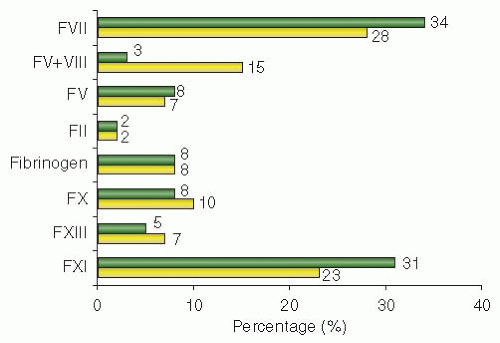
5 The WFH database, which started in 1998 but collected information since 2004, includes information from 108 countries (2008 update), while the RBDD, developed in 2004, reports data from 68 countries. According to these registries, FXI and FVII deficiencies are the most prevalent RICD worldwide, with frequencies of 29% and 33% of the total RICDs, followed by FX (9%), fibrinogen (8%), FV (8%), FXIII (6%), and combined FV+FVIII (5%) and FII deficiencies (2%) (
FIGURE 53.1).
6 These results were also confirmed by smaller national registries, such as those of Central and South America, Egypt, France, India, Iran, and the United Kingdom.
There are considerable variations in bleeding patterns among patients with RICDs, but on the whole, bleeding manifestations affecting critical organs such as the central nervous and musculoskeletal systems appear to be less frequent than in hemophilia A and B.
7 The laboratory diagnosis of RICDs is carried out by means of screening tests such as the activated partial thromboplastin time (APTT), the prothrombin time (PT), and the thrombin time (TT) in subjects reporting a clinical and family history of bleeding (
Table 53.1).
4 Abnormal results of screening tests should be followed by coagulation factor assays, in order to specify the diagnosis. Molecular diagnosis is based on the search of mutations in the genes encoding the corresponding coagulation factors. Exceptions are the combined deficiency of FV and FVIII, caused by mutations in genes encoding proteins involved in the intracellular transport of FV and FVIII (multiple coagulation factor deficiency 2 [MCFD2] and lectin mannose binding protein 1 [LMAN1]), and the combined deficiency of vitamin K-dependent proteins (FII, FVII, FIX, and FX), caused by mutations in genes that encode enzymes involved in posttranslational modifications and in vitamin K metabolism (
γ-glutamyl carboxylase [GGCX] and vitamin K epoxide reductase [VKOR]). Genetic analysis is important mainly for prenatal and preimplantation genetic diagnosis, especially in countries with a high rate of RICDs and consanguinity.
8 Because it is not always clear whether or not the identified mutation is the cause of the deficiency of a clotting factor, family history associated with allele frequency in the general population is important to rule out that the identified mutations are common polymorphisms.
7Treatment of RICDs is a difficult task, because information on the clinical management of RICDs is often scarce and replacement therapy of coagulation factors may require unlicensed products that are not readily available.
9,10 The patients’ personal and family history of bleeding are important guides for management.
9 Dosages and frequency of replacement therapy with products containing the deficient factor depend on the minimal hemostatic level of the factor, its plasma half-life, and the type of bleeding episode. Unfortunately, information on the safety and efficacy of the few available products is scarce and experiences from their use are significantly more limited than those available for congenital hemophilia. Other treatments in RICDs are nontransfusional adjuvant drugs (i.e., antifibrinolytic amino acids, estrogen-progestogen).
10After these general statements on RICD, the next section will deal specifically with each factor deficiency.
FIBRINOGEN DEFICIENCY
Fibrinogen is a 340 kDa protein synthesized in the liver, with a plasma concentration of approximately 1.5 to 3.5 g/L and a half-life of approximately 4 days (
Table 53.1). The fibrinogen molecule is a homodimer, each half consisting of the three nonidentical polypeptide chains A
α, B
β, and
γ. The genes encoding fibrinogen B
β (
FGB), A
α (
FGA), and
γ (
FGG), ordered from centromere to telomere, are clustered on chromosome 4 in a region of approximately 50 kb (
Table 53.1). Fibrin is produced by proteolytic cleavage of fibrinogen by thrombin, with the release of fibrinopeptides A and B and generation of insoluble fibrin monomer followed by polymerization. Fibrinogen is also important in primary hemostasis through its ability to support normal platelet aggregation.
9Fibrinogen deficiency is heterogeneous and two main phenotypes can be distinguished. In some cases, plasma and platelet levels of the protein are not measurable or very low, leading to afibrinogenemia and hypofibrinogenemia. In other cases,
low clottable fibrinogen contrasts with normal or moderately reduced fibrinogen antigen, causing dysfibrinogenemia and hypodysfibrinogenemia.
3
Mutation Analysis
A homozygous deletion of approximately 11 kb of the
FGA as a cause of inherited afibrinogenemia was for the first time identified by Neerman-Arbez et al.
11 in four members of a Swiss family. Since then many other mutations have been identified in afibrinogenemia (in homozygosity or in compound heterozygosity) or hypofibrinogenemia.
8 While the majority of mutations are in the
FGA gene, missense mutations leading to complete fibrinogen deficiency appear to be clustered in the highly conserved
C-terminal globular domains of the B
β and
γ chains. Recent functional studies of these mutations in transfected cells have demonstrated either impaired assembly or secretion of the fibrinogen hexamer. To date, only two missense mutations located at the start of the coiled coil in
FGA (Cys64Phe and Met70Arg) were found in fibrinogen deficiency.
12,13Besides the large number of rare, individual mutations identified in congenital afibrinogenemia, two common mutations were found in individuals of European origin, both in
FGA: the Asp153SerfsX4 mutation and the
FGA 11-kb deletion, both on multiple haplotypes. Two further residues of interest in screening for dysfibrinogenemia are residue Arg16 in exon 2 on
FGA, which is part of the thrombin cleavage site in the fibrinogen A
α chain, and residue Arg275 in exon 8 on
FGG, which is important for fibrin polymerization.
14
Clinical Phenotype and Treatment
Whereas the majority of patients with dysfibrinogenemia do not bleed at all, afibrinogenemic patients have a bleeding tendency that may manifest in the neonatal period, with 85% of cases presenting with umbilical cord bleeding.
15 Bleeding may also occur in the skin, gastrointestinal tract, or central nervous system, while recurrent joint and muscle bleeding leading to persistent damage to the musculoskeletal system is less frequent. Clinically milder symptoms such as epistaxis and menorrhagia are also frequent. First trimester abortion is common in afibrinogenemic women, less common in dysfibrinogenemia, and not reported so far in hypofibrinogenemia.
16,17 Postpartum bleeding is relatively frequent when no prophylactic replacement therapy is given. On the whole, bleeding problems are often not dramatic in patients with afibrinogenemia, despite the fact that screening coagulation tests (PT, APTT) usually result in no clot formation and that the skin bleeding time is prolonged.
2Both arterial and venous thromboembolic complications have been reported in afibrinogenemic patients, in spite of the fact that in the majority of patients no traditional thrombotic risk factors were present. One possible but as yet unproven explanation for this predisposition is that even in the absence of fibrinogen platelet aggregation does occur due to the action of von Willebrand factor. Moreover, in contrast to hemophiliacs, afibrinogenemic patients are able to generate thrombin, both in the initial phase of limited production and in the secondary burst of greater thrombin generation.
2Pertaining to replacement therapy, four products are available: fresh-frozen plasma (FFP), cryoprecipitate, virus-inactivated FFP, and fibrinogen concentrate. Safety commands avoidance of the first two if possible, because they are not virus inactivated. The latter is the treatment of choice, because it is virally inactivated and thus safer than cryoprecipitate or FFP and does not cause volume overload.
18,19 The conventional mode of treatment delivery is episodic and on demand, with fibrinogen administered as soon as possible after onset of bleeding. Long-term secondary prophylaxis (made possible thanks to the relatively long half-life of fibrinogen) with administration of fibrinogen every 7 to 14 days (particularly after central nervous system hemorrhages) has been proposed (
Table 53.2). Prophylactic therapy also appears to be necessary for successful completion of pregnancy: fibrinogen replacement must begin at least at 5 weeks gestation to prevent abortion and to maintain fibrinogen
levels at >1.0 g/L throughout pregnancy (a level of >1.5 g/L is recommended for delivery).
17
PROTHROMBIN DEFICIENCY
Prothrombin (FII) is a 72 kDa single-chain glycoprotein synthesized by the liver. It is one of the vitamin K-dependent coagulation factors and requires posttranslational carboxylation to become functionally active. Prothrombin consists of four domains—the Gla domain, kringle 1 and kringle 2 domains, and a serine protease domain—and is encoded by a gene of approximately 21 kb located on chromosome 11. FXa activates prothrombin on the surface of platelets in the presence of FV and calcium. During prothrombin cleavage, the activation peptide fragment 1 + 2 is released.
20Prothrombin deficiency is probably the rarest inherited bleeding disorder, with an estimated prevalence of 1:2,000,000 in the general population.
4 Based on the measurement of prothrombin (FII) activity and antigen level in plasma, two main phenotypes are distinguished: hypoprothrombinemia (both levels are concomitantly low) and dysprothrombinemia (normal or near normal synthesis of a dysfunctional protein).
20 No living patient with undetectable plasma prothrombin has been reported so far, consistent with the demonstration that in mice complete prothrombin deficiency by gene knockout is incompatible with life, resulting in embryonic or neonatal lethality.
21
Mutation Analysis
Genetic variants were identified all along the prothrombin gene, although frequently they involve the catalytic site, consistent with the fact that they affect the enzymatic activity of the protein.
22 Mutations in different exons may cause similar phenotypic expression of prothrombin. Most of the mutant prothrombins derive from missense mutations (80%), although insertion/deletions (10%), nonsense (6%), and splice-site (4%) mutations have also been described. The abnormalities may be classified according to the site of the defects: (a) in the activation mechanism or (b) in the protease activity of thrombin. The latter can in turn be subdivided into two subtypes: one in which there is defective amidolytic activity and another in which there is a defective interaction of thrombin with substrates because of mutations in the molecular recognition domains.
23 Other mutations indirectly affect the catalytic activity of thrombin, by perturbating of the Na
+ binding site of the enzyme. However, while
in vitro Na
+ binding seems to play a major role in thrombin activity, the natural mutant prothrombins with altered Na
+ binding described so far (Arg517Gln and Lys556Thr, in exon 13 and 14, respectively) had only a mild hemorrhagic phenotype.
24 Recent biochemical characterization of the mutants Des-Lys9/10 and Phe299(7)Val revealed an important role of the A-chain for a correct function of the catalytic B-chain of thrombin. Perhaps these natural mutations affect the interaction of divalent metals with thrombin, recently localized at the interface between the A- and B-chain that seem to stabilize the active conformation of thrombin.
25,26
Clinical Phenotype and Treatment
Clinically, heterozygotes with prothrombin deficiency are usually asymptomatic, while homozygotes with activity levels <10% of normal experience severe bleeding manifestations, such as spontaneous hematomas and hemarthroses, intracerebral bleeding, and gastrointestinal hemorrhages. In homozygous women, menorrhagia is frequent.
24,27Patients with a prothrombin coagulant activity above 20% usually do not require replacement therapy, but antifibrinolytic agents may be considered.
28 Replacement therapy is needed only in homozygous patients, in case of bleeding or to ensure adequate prophylaxis of bleeding before surgical procedures. Since no prothrombin concentrate exists, prothrombin complex concentrates (PCCs) are used to treat patients (
Table 53.2),
20 because in several clinical settings, higher levels of prothrombin may be achieved with PCCs without the harm of potential volume overload induced by FFP. However, all PCCs contain other vitamin K-dependent coagulation factors, which increase unnecessarily in plasma of patients with normal levels and could potentially cause thrombotic complications.
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements



