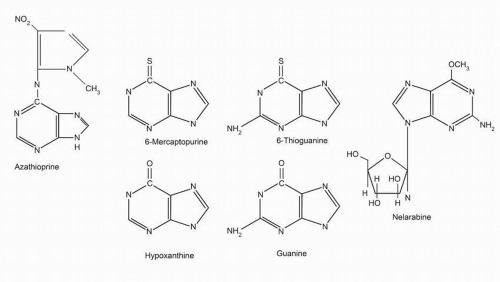
6-Mercaptopurine
6-MP is commercially available in 50-mg tablets, which also contain the inactive ingredients corn and potato starch, lactose, magnesium stearate, and stearic acid. An intravenous preparation of 6-MP has been formulated for research purposes. 6-MP is relatively insoluble and unstable in alkaline solutions.
Plasma 6-MP, 6-TG, and metabolite concentrations as low as 0.1 μM can be measured using high-performance liquid chromatography techniques.
11 Accurate kinetics of oral and intravenous preparations have been determined. Following oral administration of commonly used 6-MP doses (75 mg/m
2), peak plasma concentrations of 0.3 to 1.8 μM are seen within a mean of 2.2 hours.
12 The volume of distribution exceeds that of total body water (0.9 L/kg). There is little penetration into the cerebrospinal fluid (CSF). With high-dose oral 6-MP (500 mg/m
2), plasma 6-MP concentrations of 5 to 12 μM are achieved.
13 In human leukemic cell culture models, concentrations of 1 to 10 μM are cytotoxic. Following intravenous dosing, the half-life of 6-MP is 50 to 100 minutes and plasma concentrations of 6-MP reach 25 μM and CSF concentrations 3.8 μM.
14 Only weak protein binding is noted with 6-MP (20% bound).
Oral absorption of 6-MP is incomplete and highly variable.
15 At a dose of 75 mg/m
2, 6-MP, mean 6-MP bioavailability is 16% (range, 5%
to 37%).
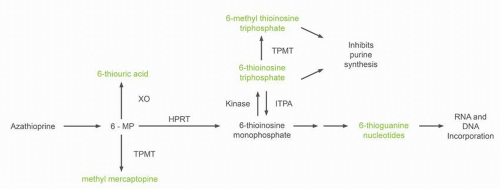
16 Clearance occurs primarily through two routes of metabolism. 6-MP is oxidized to the inactive metabolite, 6-thiouric acid, by xanthine oxidase (
Fig. 11-2). 6-MP also undergoes S-methylation by the enzyme, thiopurine methyltransferase (TPMT), to yield 6-methyl mercaptopurine. The intestinal mucosa and liver contain high concentrations of the enzyme xanthine oxidase. The low bioavailability of 6-MP is the result of a large first-pass effect as drug is absorbed through the intestinal wall into the portal circulation and metabolized by xanthine oxidase. The use of concomitant allopurinol (an inhibitor of xanthine oxidase) increases 6-MP bioavailability fivefold.
17 Allopurinol does not alter the plasma kinetics of intravenously administered 6-MP, although more 6-MP and
less thiouric acid are excreted in the urine following allopurinol therapy.
18 Methotrexate, often used with 6-MP in maintenance treatment of ALL, is a weak inhibitor of xanthine oxidase. Concomitant use of methotrexate results in a small increase in the bioavailability of 6-MP. However, the modest increase in bioavailability is thought not to be clinically significant.
15 The plasma concentration versus time profile of 6-MP differs in the same patient when studied on repeated occasions.
19 High-dose, oral 6-MP (500 mg/m
2) has been used in an attempt to saturate the first-pass metabolism of 6-MP, thereby increasing bioavailability.
13 Even at a dose of 500 mg/m
2 6-MP, xanthine oxidase is not saturated and no improvement in bioavailability is seen. Food intake and oral antibiotics reduce the oral absorption of 6-MP.
20,
21 The drug should be taken 1 to 2 hours after eating.
As previously mentioned, two catabolic pathways for 6-MP metabolism exist that significantly affect drug activity: one via xanthine oxidase (just discussed) and a second via TPMT. Patient-to-patient variation in TPMT activity results in significant changes in 6-MP metabolism and drug toxicity. As seen in
Figure 11-2, TPMT catalyzes the S-methylation of 6-MP to a relatively inactive metabolite, 6-methyl mercaptopurine (6CH
3MP). TMPT also metabolizes thioinosine monophosphate into methyl thioinosine monophosphate, a molecule that can inhibit de novo purine biosynthesis. The frequency distribution of TPMT activity in large population studies is trimodal.
22 One in two hundred to three hundred subjects has absent enzyme activity; 10% of the population has intermediate activity, and the rest have high enzyme activity. A reciprocal relationship between TPMT activity and the formation of 6-thiopurine nucleotides has been demonstrated. Over 20 nonsynonymous variations in the TMPT gene have been identified, 17 of which have reduced TMPT activity.
23 Among these genetic variations, three (TPMT
*2, TPMT
*3A, and TMPT
*3C) account for 90% of patients with low or intermediate TMPT activity. The proteins formed by TPMT
*2 and TPMT
*3A have degradation half-lives of 15 minutes compared to a half-life of 18 hours for wild type TPMT.
24 Lymphoblasts from individuals homozygous or heterozygous for a variant TMTP gene have lower TMPT activity than lymphoblasts homozygous for the normal gene.
25 Patients with low TPMT activity are susceptible to 6-MP and 6-TG-induced myelosuppression. Genetic testing using PCR-based methods can now identify TPMT-deficient and heterozygous patients.
26 This test, as opposed to direct measurement of TPMT activity in red blood cells, is not affected by prior blood transfusions to the patient. In 2004, the FDA suggested TMPT testing for patients developing myelosuppression with standard dose 6-MP with subsequent dosage modifications for TMPTdeficient patients.
27 Patients with absence of TMPT activity should have the dose of 6-MP reduced, but not the dose of other chemotherapeutic agents. It has been suggested that children with ALL should receive only 5% to 10% of a standard mercaptopurine dose
if they are homozygous for a variant TMPT gene and 65% if they are heterozygous.
28Several studies,
29,
30 but not all,
16 have suggested that children with high TPMT activity are at greater risk of disease relapse as a result of decreased drug activation. In a large Scandinavian study in children with ALL, the risk of relapse following therapy was 18% for patients with wild type TPMT versus 6% for patients heterozygous or deficient in TMPT activity (
P = 0.03).
31 Despite a lower probability of relapse, patients with low TPMT activity did not have superior survival (
P = 0.08), perhaps due to an increased rate of drug toxicity including the development of secondary malignancies.
While screening for TMPT variants with decreased enzymatic activity identifies individuals at risk for mercaptopurine toxicity, the association is not perfect. Toxicity still occurs in individuals with wild-type TMPT. Individuals heterozygous for TPMT
*2,
*3A and
*3C can have a wide range of TPMT activity. Mercaptopurine metabolism is not dependent on a single gene (
Fig. 11-2). Inosine triphosphate pyrophosphatase (ITPA) catalyzes the hydrolysis of inosine triphosphate (ITP) to inosine monophosphate which protects cells from the accumulation of potentially harmful nucleotides. A polymorphism in the ITPA gene occurring in roughly 10% of individuals results in a 25% decrease in enzyme activity in heterozygous individuals.
32 ITPA inactivates the potentially toxic thioinosine triphosphate metabolite from patients receiving mercaptopurine or azathioprine (
Fig. 11-2). In children with ALL who have had their mercaptopurine dose adjusted based on TMPT phenotype, the ITPA mutant allele has been associated with an increase incidence of febrile neutropenia
33 and myelosuppression.
34 However, an association of ITPA polymorphism with drug toxicity has not been seen in all studies.
32
6-Thioguanine
6-TG is available as 40-mg tablets for oral use. An intravenous preparation is investigational. As with 6-MP, the absorption of 6-TG in humans is variable and incomplete (mean bioavailability is 30%; range: 14% to 46%).
35 Peak plasma levels of 0.03 to 5 μM occur 2 to 4 hours after ingestion; the median drug half-life is 90 minutes but with wide variability reported.
36 Intravenously administered 6-TG has been evaluated. Clearance of drug (600 to 1,000 mL/min/m
2) appears to be dose dependent, suggesting saturation of clearance at doses over 10 mg/m
2/h.
37 Plasma concentrations of 4 to 10 μM can be achieved.
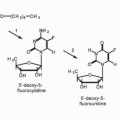
The catabolism of 6-TG differs from that of 6-MP. Thioguanine is not a substrate for xanthine oxidase. Thioguanine is converted to 6-thioinosine (an inactive metabolite) by the action of the enzyme, guanase. Because thioguanine inactivation does not depend on the action of xanthine oxidase, allopurinol will not block the detoxification of thioguanine. In humans, methylation of thioguanine, via TPMT, is more extensive than methylation of 6-MP. The product of methylation, 2-amino-6-methylthiopurine, is substantially less active and less toxic than thioguanine.