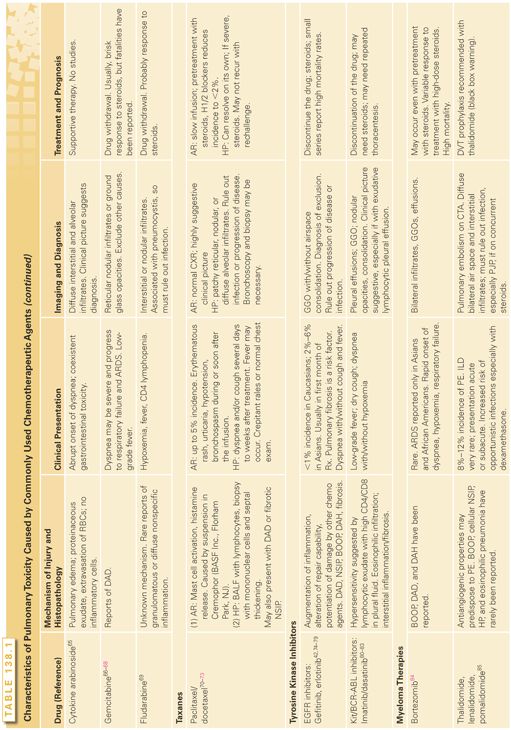
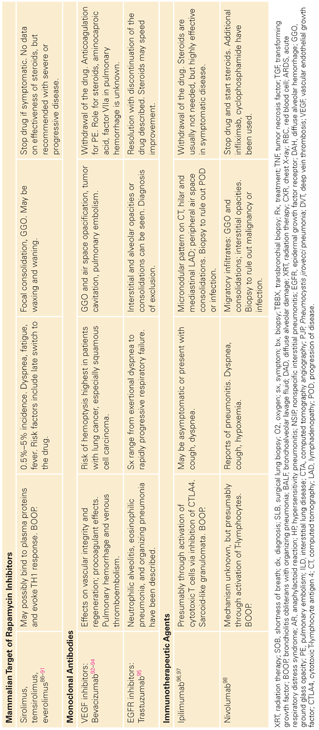
Mechanisms of Pulmonary Injury and Histology
Except for a few chemotherapeutic agents (e.g., bleomycin), the details of the pathophysiology of lung injury are unknown. Various mechanisms of pulmonary toxicity have been proposed based on the mechanisms of action of different classes of therapeutic agents. These include a direct toxic effect on alveolar epithelial cells, the induction of an inflammatory immunologic response and endothelial cell injury, or activation causing capillary leak syndrome. These events can result in a variety of clinical presentations including nonspecific interstitial pneumonitis, hypersensitivity reaction syndrome, noncardiogenic pulmonary edema, infusion reaction, capillary leak syndrome, eosinophilic pneumonia, bronchospasm, acute lung injury, diffuse alveolar damage, or fibrosis.
The histopathologic changes of drug-induced pulmonary toxicity show common features. Similar to radiation-induced damage, abnormalities are seen in endothelial and epithelial cells. The vascular damage is characterized by endothelial swelling with exudation of fluid into the interstitium and the intra-alveolar spaces. Destruction and desquamation of type I pneumocytes occurs with delamellation and proliferation of type II pneumocytes. Mononuclear cell infiltration and fibroblast proliferation with fibrosis are common findings; the character of the inflammatory cellular infiltrate may be a feature that distinguishes the toxicity of one drug from another.
Pathways involved in chemotherapy-induced lung disease include apoptosis, impaired repair response, and angiogenesis. Apoptosis pathways include the death receptor pathway and the Fas ligand as well as the mitochondrial pathway and Bcl-2 protein. Both of these lead to activation of caspases, which activate degradative enzymes within the cell. Cells affected can be immune cells, endothelial cells, or epithelial and other parenchymal cells. Impaired pathways involving epidermal growth factor and TGF may contribute to alterations in repair mechanisms; both factors are expressed in type II pneumocytes and epithelial cells.
The older chemotherapy agents are the best described in their mechanism of lung injury. It is thought that a major contributor to the toxicity from these agents results from ROS including superoxide anions, hydrogen peroxide, and hydroxyl radicals, primarily from activated neutrophils.105 Bleomycin induces reactive oxygen radicals by forming a complex with Fe3+. Consistent with a direct pathologic role for this mechanism, iron chelators ameliorate the pulmonary toxicity of bleomycin in animal models.45 ROS formed after bleomycin therapy can produce direct toxicity through participation in oxidation-reduction reactions and subsequent fatty acid oxidation, which leads to membrane instability.46 Oxidants also cause other inflammatory reactions within the lung. For example, the oxidation of arachidonic acid is an initial step in the metabolic cascade that produces active mediators, including prostaglandins and leukotrienes.47 Because ROS are so highly tied to bleomycin toxicity, high-dose oxygen can exacerbate its effects, and toxicity can be seen years after its administration.48,49 In addition to ROS direct toxicity, cytokines such as IL-1, macrophage inflammatory protein-1, monocyte chemoattractant protein-1, and TGF-β are released from alveolar macrophages in animal models of bleomycin toxicity resulting in fibrosis.50,51,105 Through modulation of fibroblast proliferation, bleomycin can cause excessive interstitial and intra-alveolar collagen deposition leading to pulmonary fibrosis.51 Bleomycin directly upregulates collagen synthesis in fibroblasts by stimulating transcription. There is some evidence to support a role for a mast cell/fibroblast interaction in the generation of this fibrosis as well.53
One of the potential determinants of bleomycin toxicity is the bleomycin hydrolase, which is the major enzyme responsible for metabolizing bleomycin to a nontoxic molecule.54 Interestingly, the two organs that are the most common targets for bleomycin toxicity (lung and skin) have the lowest levels of the enzyme. Studies are needed to determine if genetic variability of this enzyme accounts for individual susceptibility or immunity to bleomycin pulmonary toxicity in humans, as it does in animals.55–57 Likewise, with linkage and association studies, genetic susceptibility to bleomycin-induced pulmonary fibrosis has been investigated in mice. Polymorphisms in Cep55, a gene encoding proteins involved in autophagy, and Masp2, a gene encoding complement pathway proteins, as well as several others, have been identified as potentially increasing toxicity susceptibility.58,59 However, these associations have not been replicated in other studies and are currently not being used clinically.60
Because the lung is exposed to many substances that can activate its immune system, there appears to be a pulmonary immune tolerance state to avoid overreactions. This tolerance state in part may be a result of an effector and suppressor cell balance. Cytotoxic drugs can alter the normal balance, which may cause tissue damage.41 For example, lymphocytic alveolitis is a consistent finding in methotrexate pneumonitis, with an imbalance of the CD4-to-CD8 ratio.106 Bronchoalveolar lavage studies in patients with methotrexate pulmonary toxicity have shown the presence of a T-lymphocytic alveolitis, whereas studies on some patients with bleomycin toxicity have revealed a polymorphonuclear alveolitis.106–107 G-CSF is also implicated in pulmonary toxicity through its effects on neutrophil activation. G-CSF toxicity is thought to be secondary to activation of inflammatory pathways and ROS within the neutrophils, causing pulmonary edema and acute respiratory distress syndrome.108
IL-2, used to treat metastatic melanoma and renal cell cancer, stimulates the immune system by stimulating CD4+ T cells and increasing natural killer cell activity. These effects likely contribute to a generalized inflammatory state, contributing to the capillary leak syndrome and noncardiogenic pulmonary edema associated with this drug. IL-2 has been shown to directly increase endothelial permeability and indirectly increases vascular leak by increasing cytokine levels, particularly tumor necrosis factor (TNF)-α, leukocyte adhesion, and inflammation, and altering the extracellular matrix.109
Tyrosine kinase inhibitors, although generally well tolerated, can show serious pulmonary toxicity. Epidermal growth factor receptor (EGFR) is a target of these newer molecular-targeted chemotherapies including gefitinib and erlotinib. Inhibition of the EGFR pathway can exacerbate damage from other chemotherapy agents or sepsis by altering the repair capability of cells. It has been shown that inhibition of the EGFR pathway can upregulate genes that promote prolonged inflammation.110 Angiogenesis, which involves VEGF, TNF-α, TGF-β, and platelet-derived growth factor, also are targets of tyrosine kinase inhibitors, and impaired function of these factors likely also contribute to pulmonary toxicity. Inhibition of the platelet-derived growth factor pathway, seen with dasatanib, is thought to contribute to the pathogenesis of pleural effusions, which is seen in up to 54% of patients treated with this agent. Interestingly, pulmonary hypertension can also be seen with this agent, although the mechanism is unclear. Other Abelson gene inhibitors do not show this effect.111 Dasatinib, axitinib, crizotinib, pazopanib, and sunitinib have been reported to cause pulmonary embolism.112
Mammalian target of rapamycin inhibitors, such as everolimus and sirolimus, which inhibit cell proliferation, growth, and angiogenesis by inhibiting phosphatidylinositol kinases, specifically mTORC1, also are thought to cause a pneumonitis by a T-cell–mediated delayed-type hypersensitivity.87 In bronchoalveolar lavage (BAL) fluid of patients with pneumonitis from sirolimus, a CD4+ T-cell lymphocytosis is found. Re-exposure to sirolimus may result in antigen presentation and the activation of Th1-cells and recruitment of macrophages and production of proinflammatory cytokines.87,113
Bortezomib, thalodimide, and lenolinomide are all used to treat multiple myeloma. Bortezomib, a proteosome inhibitor, has been associated with parenchymal changes, organizing pneumonia, and pleural effusions; thalodimide, which downregulates TNF-α and has antiangiogenic properties, is associated with pulmonary embolism, organizing pneumonia, and pneumonitis.114,115 The mechanism of pulmonary toxicity associated with bortezomib and thalidomide is unclear.
Bevacizumab and other anti-VEGF therapies can cause epistaxis. Pulmonary hemorrhage and pulmonary embolism have been reported. The mechanism of hemorrhage and thrombosis likely reflects the multiple actions of VEGF on vasculature and on the coagulation system. Inhibition of VEGF reduces regeneration of endothelial cells and can cause defects that expose procoagulant phospholipids on the luminal plasma membrane or underlying matrix, leading to thrombosis or hemorrhage.116 VEGF also increases production of nitric oxide and prostacyclin (prostaglandin I2); suppresses pathways involved in endothelial cell activation, apoptosis, and procoagulant changes; and inhibits proliferation of vascular smooth muscle cells.117 Reduction in nitric oxide and prostaglandin I2 by anti-VEGF agents may predispose to thromboembolic events. A few cases of pulmonary arterial hypertension have been reported with bevacizumab.85
Clinical Features
Combination chemotherapy, concurrent radiation treatments, oxygen therapy, renal dysfunction, preexisting pulmonary disease such as idiopathic pulmonary fibrosis, chronic obstructive pulmonary disease, radiation pneumonitis, extensive pulmonary metastases, and poor functional status and advanced age have been associated with increased pulmonary toxicity. Table 138.2 lists some of the drugs that have been associated with these predisposing factors.41 Because bleomycin toxicity is relatively common, it deserves special mention. Although toxicity drastically increases with doses in excess of 450 to 500 mg, it can occur with much lower doses, especially when other risk factors are present. Renal damage after cisplatin administration, with subsequent accumulation of bleomycin, was a likely cause of pulmonary toxicity and 67% mortality reported early in its use. If possible, bleomycin should precede cisplatin infusion to minimize the risk. Although supplemental oxygen has been a classic cofactor in bleomycin pulmonary toxicity, there are no large controlled studies.41 An increased risk of pulmonary toxicity (4 of 12 patients, fatal in 3 of 4) was described in a small uncontrolled study of patients receiving G-CSF in combination with bleomycin-containing chemotherapy for non-Hodgkin’s lymphoma.118
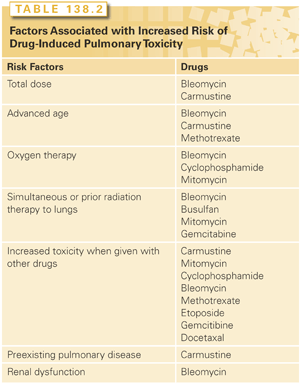
Long intervals between drug administration and onset of clinical toxicity have been described. Late-onset pulmonary fibrosis has been reported many years after cyclophosphamide and carmustine are discontinued.41
Signs and Symptoms
The cardinal symptom of drug-induced pulmonary toxicity is dyspnea; however, symptoms can range from nonproductive cough with low-grade fevers to overt respiratory failure. If bronchospasm occurs, shortness of breath is accompanied by wheeze. Fibrosis typically causes basilar crackles, although clubbing is not found. Increased fremitus and decreased breath sounds are signs of pleural effusion. Other characteristics of chemotherapy-induced pulmonary disease are outlined in Table 138.1. These findings can occur immediately following administration of the drug or over a treatment cycle. Fibrosis may take years to develop. Chest pain has been reported during infusion of bleomycin or immediately after therapy with methotrexate; however, it is an unusual manifestation of toxicity.41,55 A syndrome of acute dyspnea, probably resulting from direct toxicity to the pulmonary vasculature, can occur during or shortly after vinca alkaloid infusion when given in combination with mitomycin for treatment of nonsmall-cell lung cancer.61 Because hemoptysis is an uncommon feature of drug-induced pulmonary toxicity unless treated with an anti-VEGF therapy, when it is present, other diagnoses should be considered.
Capillary leak syndrome can lead to a variety of symptoms. These include pulmonary edema that will manifest as tachypnea, low pulse oximetry readings, or in severe cases which lead to mechanical ventilation, cardiovascular symptoms that include tachycardia and hypotension or shock as well as renal insufficiency and hepatic insufficiency. For IL-2 therapy specifically, weight gain from loss of albumin can be seen after 3 to 4 days of administration of the drug and typically resolve after 2 weeks. However, manifestations can appear later and take longer to resolve as well.
All-trans-retinoic acid treatment of leukemias can induce the retinoic acid syndrome, which consists of fever, dyspnea, weight gain, pulmonary infiltrates, pleural or pericardial effusions, hypotension, renal dysfunction, and leukocytosis.119 The pulmonary disorder is thought to be mediated by newly differentiated leukemia cells marginating into the pulmonary circulation, increasing capillary permeability and releasing cytokines that induce neutrophil migration into the interstitium. High doses of corticosteroids are the most effective treatment, and steroid prophylaxis has been reported to be useful.
Diagnostic Imaging
The most common radiographic abnormality associated with drug-induced pulmonary toxicity is a reticulonodular or interstitial pattern with thickened septal lines or both, which may be basilar or diffuse. These changes can be seen with many different chemotherapies including antimetabolites, taxanes, and molecular-targeted therapies. Pleural effusions can be seen alone, as with tyrosine kinase inhibitors or thalidomide, or in the setting of heart failure or capillary leak syndrome, which is described with IL-2 therapy, G-CSF, and gemcitabine. Distinguishing capillary leak from heart failure is important as it will affect ongoing therapy and response to steroids. Effusions are also found with treatment from mitomycin, busulfan, methotrexate, and procarbazine toxicity.
EGFR inhibitors are associated with GGOs, which should be distinguished from tumor progression. They can cause dense consolidation with associated GGOs within 1 week to 1 month after initiation of therapy. Other CT patterns include patchy GGOs with interlobular septal thickening, or extensive bilateral GGOs with consolidation and traction bronchiectasis. The latter pattern carries the highest mortality rate.
Bleomycin pulmonary toxicity can have several patterns on chest CT, notably interstitial pneumonitis, cryptogenic organizing pneumonia, or fibrosis. Cavitating and noncavitating nodules, simulating metastatic disease, have been seen with bleomycin toxicity as well.41
Methotrexate toxicity often presents with bilateral interstitial and alveolar infiltrates along with pleural effusions. Hilar adenopathy is distinctly unusual in chemotherapy toxicity but has been reported with methotrexate toxicity.41 These changes can be seen with fever and eosinophilia and will usually not progress to fibrosis if the medication is stopped. These findings typically occur within the first year of therapy.
In some instances, the chest radiograph is normal, even in the presence of histologically proven pulmonary infiltration and fibrosis.41 Most commonly, methotrexate and carmustine toxicity have been reported, with normal chest radiographic findings. Finally, rituximab can cause interstitial infiltrates with cryptogenic organizing pneumonia in the setting of rapid onset of fever, dyspnea, and cough; however, this is a rare finding. Other CT findings of toxicity are GGO associated with vasculature that proves to be pulmonary hemorrhage. This is most often seen with bevacizumab.
Pulmonary Function Tests
The most common abnormalities associated with chemotherapy-induced pulmonary toxicity are a reduced diffusing capacity for carbon monoxide and a restrictive ventilatory defect.41
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


