The Ubiquitin-Proteasome Pathway
The UPP has a major role in regulating a broad number of fundamental cellular pathways including apoptosis,
33 cell growth and
proliferation,
34 DNA repair,
35 unfolded protein response (UPR),
33,
36 and immune response.
37 Alterations in these pathways have been implicated in multiple diseases, particularly cancer. Therefore, compounds interfering with proteasome functions may have a major impact on cancer cell fate.
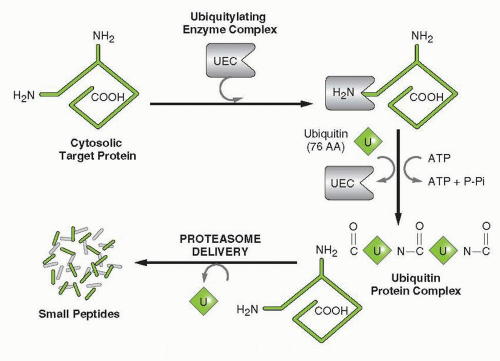
Broadly speaking, the UPP consists of two major components: the ubiquitinating enzyme complex (UEC) and the degradation system (26S proteasome). Three enzymes make up the UEC, the ubiquitinactivating enzyme (E1), the ubiquitin-conjugating enzyme (E2), and the ubiquitin-conjugating ligase (E3). E1 is a generic enzyme used
by the pathways regardless of the protein substrate targeted. By contrast, there are 20 to 30 different ubiquitin-conjugating enzymes (E2) and likely hundreds of E3 ligases. The ligase steps are the points where the specificity of the ubiquitylation process is controlled, with most protein substrates having their own distinct ligase.
38,
39 Each of these enzymes represents a potential therapeutic target offering unique ways to specifically inhibit the degradation of a very selective protein or groups of proteins. The initial step involves the binding of the UEC to the N-terminus of the target (
Fig. 21-2). This enzyme complex catalyzes the covalent linkage of ubiquitin molecules to the ε-amino moieties of internal lysine residues in a processive manner. These ATP-dependent catalytic reactions eventually lead to the generation of a branched polyubiquitylated protein. This ubiquitylation process is the primary means by which the cells “tag” or “earmark” specific proteins for degradation at the 26S proteasome.
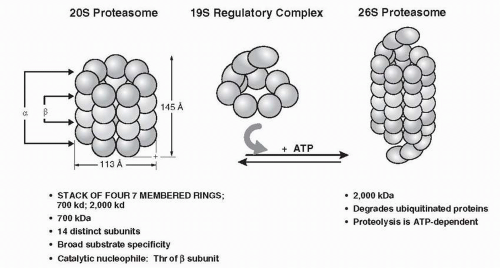
The second major component of the pathway includes the proteasome (
Fig. 21-3), which is composed of two structures, the 20S proteasome and the 19S regulatory subunit. Collectively they form a complex called the 26S proteasome.
40 The isolated 20S proteasome does not exhibit proteolytic activity in the absence of the 19S regulatory subunit. The 19S subunit mediates the ATP-dependent process of denaturing and unfolding the ubiquitin conjugates. The 26S proteasome is a large structure with highly processive threonine proteolytic activity, and as a result it cleaves polypeptides at multiple sites, releasing very small peptides ranging in length from 2 to 24 amino acids.
41,
42,
43The 19S regulatory subunit includes multiple peptidases that disassemble and unfold the polyubiquitin conjugates. It also plays a major role in regulating and facilitating the multiple ATP-dependent processes used by the proteasome including 20S and 19S subunits assembly, protein unfolding, ubiquitylation, opening of the regulatory “gate” that allows the protein to enter the 26S lumen, and the action of the isopeptidases leading to the recycling of the ubiquitin molecules.
44The 20S proteasome consists of four rings, each containing seven individual globular proteins. As shown in
Figure 21-3, the assembly of the rings forms a central lumen through which proteins are funneled. The two outer layers of the 20S proteasome are the α-layers that allow anchorage of the 19S regulatory subunit. The two inner rings are the
β-layers containing the proteolytic activity of the proteasome. The 19S subunit sits on top and bottom of the 20S subunit, controlling the entry of ubiquitylated proteins into the core of the proteasome.
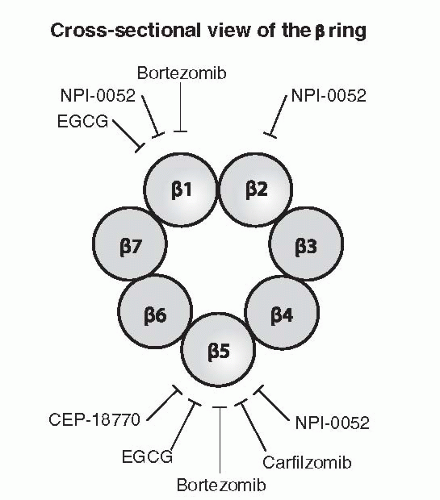
44 Once the protein enters the lumen of the proteasome, the different proteases digest the protein into smaller peptides fragments. At least three different enzymatic activities have been ascribed to the
β-layers, including (1) a peptidylglutamyl activity (
β1) cleaving proteins near glutamate residues, (2) a trypticlike function (
β2) that cleaves proteins near lysine and arginine residues; and (3) a chymotryptic-like function (
β5) that cleaves proteins near phenylalanine, tyrosine, and tryptophan.
40,
45,
46,
47,
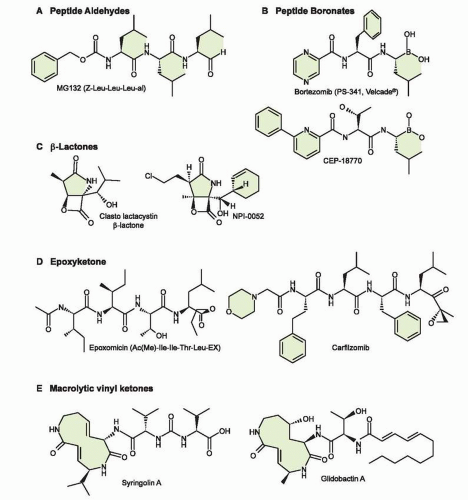
48 Figure 21-4 shows the targets of several PIs that are in the clinic including bortezomib, carfilzomib, salinosporamide A, and CEP-18770.
NF-κB, Endoplasmic Reticulum Stress, Unfolded Protein Response, and Apoptosis
One of the first observations suggesting the possibility that PIs could be used as anticancer agents was the early discovery that proteasome inhibition could lead to inactivation of NF-
κB by stabilizing I
κB and preventing the translocation of NF-
κB to the nucleus.
49,
50 NF-
κB family is composed of several members including RELA, RELB, REL (cREL), NF-
κB1, and NF-
κB2. These proteins form heterodimeric and homodimeric complexes and their activities are regulated by two pathways known as the classical or canonical pathway and the alternative or noncanonical pathway.
51 In both pathways, ubiquitylation and degradation of critical components lead to translocation of RELA-p50 dimers (canonical pathway) and REL-p52 and REL-p50 (noncanonical) to the nucleus followed by binding to target gene DNA.
52,
53,
54,
55 Abnormal regulation of NF-
κB and the signaling pathways that control its activity are involved in cancer development and progression.
56 NF-
κB activation affects all hallmarks of cancer through the transcription of genes involved in cell proliferation, angiogenesis, metastasis, inflammation, and suppression of apoptosis.
56,
57,
58,
59,
60,
61 Constitutive activation of NF-
κB has been described in several solid tumors as well as in a number of hematological malignancies including MM.
62,
63,
64 Moreover, leukemia, lymphoma, and MM cells have been reported to undergo apoptosis upon treatment with various inhibitors of NF-
κB signaling including PIs.
65,
66,
67,
68,
69 Inhibition of NF-
κB correlates with a decrease in the expression of antiapoptotic and antiproliferative genes such as BCL
XL, XIAP, cIAP2, interleukin (IL)-6, and cyclin D1.
62,
70,
71,
72 Furthermore, interaction of bone marrow stromal cells with MM cells leads to NF-
κB activation and promotes tumor growth.
73,
74 Bortezomib is approved by the FDA for the treatment of MM and its efficacy is at least in part due to inhibition of NF-
κB activity. Indeed, gene expression DNA microarray analysis has established a correlation between NF-
κB signature and clinical outcome.
75The endoplasmic reticulum (ER) is an organelle that has an essential role in multiple cellular processes that are required for cell homeostasis and survival. ER stress (ERS), a condition typically associated with accumulation of misfolded or unfolded proteins in the lumen of the ER, triggers an evolutionarily conserved response termed the unfolded protein response (UPR).
76 Cell response to UPR is first to adapt and reestablish cell homeostasis by increasing the protein folding capacity of the ER.
77,
78 The UPR-induced alarm refers to signal transduction events that are commonly associated with cellular stress, including activation of MAPKs, JNK, and p38 MAPK. In addition, kinases responsible for activation of NF-
κB may be induced.
77 Finally, when the adaptive mechanisms put in place by the UPR fail to counteract the ERS, then cell death is induced through different mechanisms.
79,
80,
81,
82 The important roles of ERS and the UPR in tumor biology make them novel therapeutic targets in cancer. Bortezomib induces ERS-mediated apoptosis in tumor cells.
83,
84,
85,
86Interestingly, ERS has also been implicated in the antitumor effects of cisplatin and geldanamycin.
87,
88,
89 Bortezomib enhances ERS induced by both compounds, resulting in increased antitumor activity in an orthotopic pancreatic cancer xenograft model.
86 Cells exposed to bortezomib form aggregates of ubiquitin-conjugated proteins, or “aggresomes,”
in vitro and
in vivo.90 Bortezomib-induced aggresome formation may serve a cytoprotective role by allowing cells to dispose of accumulated unfolded proteins that result from proteasome dysfunction
91 and could represent a mechanism of resistance to bortezomib. Notably, bortezomib-induced aggresome formation could be disrupted using histone deacetylase (HDAC) 6 small interfering RNA or chemical HDAC inhibitors.
86,
92,
93 These events result in ERS and induction of apoptosis in pancreatic cancer cells, MM, and chronic myeloid leukemia (CML).
94,
95,
96 These results provided the rationale for the development of the combination of bortezomib and vorinostat in patients with refractory and relapsed MM patients.
97 The results of this phase I trial are encouraging and have prompted further development of the combination in other tumor histologies including non-Hodgkin’s lymphoma (NHL), soft tissue sarcoma, and glioblastoma multiforme. ERS is probably related to the strong activity of bortezomib in MM patients where highly secretory plasma cells may overload the ER. The preclinical and early clinical results suggest that bortezomib-mediated disruption of the UPR represents a novel strategy to enhance the antitumor activity of agents that induce cell death via a classic ERS-dependent mechanism.
Effects on Cell Cycle Regulation
Control of cell cycle transition points depends heavily on both transcriptional and posttranscriptional mechanisms. The orderly degradation of key regulatory proteins though the UPP allows for the coordinated progression of cells through the different stages of the cell cycle, mitosis and proliferation. Inhibition of the degradation of any of the key proteins that control this well-orchestrated process can have profound consequences on cell cycle kinetics and tumor growth. Treatment of human colon cancer lines with the PI lactacystin leads to cell cycle arrest mediated by p21 accumulation.
98 Another well-established cell cycle control mechanism revolves around cyclin B, the synthesis of which begins early in the S phase, accumulating during G2 and early mitosis. Progression into and through anaphase is dependent on the degradation of cyclin B, which is regulated through ubiquitylation and proteolytic degradation by the proteasome.
99 Similarly, progression to S phase from G1 is regulated by the cyclin E-CDK2 complex. Association of this complex with p27 is responsible for inhibiting the kinase activity leading to cell cycle arrest.
100,
101 P27 is also a target of the proteasome.
100,
102,
103 Therefore, a potential important mechanism of action of PIs is to disrupt normal regulation of cell cycle kinetics. The relevance of these findings is not completely clear; however, studies have shown that most MCLs have a decreased or lost expression of p27 with normal p27 mRNA.
104,
105 Furthermore, overexpression of p53 and lost expression of p27 in patients with MCL correlate with a significant reduction in overall survival.
104
Pharmacology
Preclinical models have extensively demonstrated that PIs have potent anticancer activities. Lactacystin induces cell death in leukemia cells at concentration of about 5 μM,
67 and bortezomib inhibits
tumor growth in a broad variety of tumor models.
106,
107,
108 Normal cells were found to be more resistant to PIs than tumor cells.
109,
110 Furthermore, within the dose range of bortezomib that induces tumor growth inhibition in xenograft models, normal hematopoietic cells seem not to be significantly affected by the cytotoxic effects of bortezomib. Boronate PIs have been shown to kill tumor cells in culture, as demonstrated by their activity in the tumor cell line screen.
111 Data from the NCI’s algorithm COMPARE demonstrated that the mechanism of cytotoxicity of bortezomib was strikingly different from any of the other 60,000 compounds in the library. The average growth inhibition for bortezomib was 7 nM across the entire panel of cells. In prostate cancer cell lines, bortezomib induces G2-M growth arrest that was associated with the accumulation of p21 and p27. Cell death was noted at 20 nM for cells incubated for 24 hours with bortezomib.
111 Studies with radiolabeled bortezomib have shown that bortezomib is broadly distributed to all tissues in rats and cynomolgus monkeys and no detectable drug could be found crossing the blood-brain barrier.
111 In vivo models of drug-resistant human myeloma have shown that a twice weekly schedule of bortezomib administration resulted both in tumor shrinkage and prolonged median survival of treated mice.
112,
113
Clinical Pharmacology
Studies on nonhuman primates have shown that after a single intravenous dose of bortezomib, plasma concentrations decline in a classic biphasic manner with a
t1/2 α of approximately 10 minutes.
114 The terminal elimination phase in humans has been estimated between 5 and 15 hours. Multiple doses of drug appear to result in some decrease in clearance, with a resulting increase in the terminal elimination half-life (
t1/2;) and the area under the curve (AUC), but they have no effect on the estimated maximum plasma concentration (
Cmax) or distribution half-life. Similar pharmacokinetic profiles have also been observed in preclinical studies and do not appear to result in increased toxicity from accumulation of the drug with repeat dosing. The overall disposition of bortezomib is most consistent with a two-compartment pharmacokinetics (PK) model and the principal pathway of elimination is through oxidative deboronation. Based on
in vitro studies, the major phase 1 metabolic reactions are mediated by cytochrome P450 isoforms 3A4 and 2C19.
115 Bortezomib exposure is increased in the presence of ketoconazole, a potent CYP3A inhibitor. On the other hand, omeprazole, a potent inhibitor of CYP2C19, has no significant effect on the exposure of bortezomib.
Pharmacokinetics of Bortezomib in Patients with Renal and Liver Dysfunction
A study by Mulkerin et al.
116 assessed the PK and pharmacodynamic profiles of bortezomib in patients with advanced malignancies and normal, mild, moderate, or severe renal insufficiency. The study was designed to evaluate safety and tolerability, as well as to determine the maximum tolerated dose (MTD) of bortezomib in this patient population.
Exposure to bortezomib was comparable among all groups and was not affected by the degree of renal impairment.
A PK study is being conducted, by the National Cancer Institute Organ Dysfunction Group, in patients with various degree of hepatic impairment who were classified according to the NCI classification of hepatic impairment (
Table 21-2). Patients received bortezomib doses ranging from 05 to 1.3 mg/m
2. An interim analysis revealed that the exposure of bortezomib was increased in patients with moderate and severe hepatic impairment. Therefore, in this population, bortezomib should be started at lower dose and patients monitored closely.