Patrick J. Bosque, Kenneth L. Tyler
Prions and Prion Diseases of the Central Nervous System (Transmissible Neurodegenerative Diseases)
Prions are the transmissible agents of a class of neurodegenerative diseases of humans and other mammals. Prions differ from other transmissible agents of disease in that they contain no information-bearing nucleic acid. They are composed mainly, perhaps only, of abnormally folded and aggregated forms of a normally expressed protein called the prion protein (PrP). The human forms of prion disease are most commonly referred to as Creutzfeldt-Jakob disease (CJD), although some specific clinical forms carry other names, notably Gerstmann-Sträussler-Scheinker syndrome (GSS) and fatal familial insomnia (FFI). The most important prion diseases of animals are scrapie of sheep, bovine spongiform encephalopathy (BSE) of cattle, and chronic wasting disease (CWD) of deer and related species.
Scrapie is the archetypical prion disease. It was first recognized as a distinct clinical entity in 18th-century western Europe.1 Studies on scrapie in the late 19th through mid-20th century, particularly those of Cuillé and Chelle,2,3 established that the disease was transmissible by a filterable agent after incubation periods of up to 22 months. Later studies uncovered the extraordinary resistance of the scrapie agent to ionizing radiation and other physical and chemical treatments that ordinarily inactivate viruses. This led Alper and co-workers4 to suggest, in 1966, that the agent might replicate despite lacking nucleic acid.
Gajdusek and co-workers5,6 identified kuru, a neurodegenerative condition endemic in certain cannibalistic tribes in the highlands of New Guinea, as a human prion disease when they transmitted the condition to primates.7 They were aided in this discovery by the insights of the veterinary neuropathologist Hadlow, who remarked on the histopathologic similarity of kuru to scrapie, triggering the search for the potential transmissibility of kuru.8,9 The similarities between kuru and scrapie that first drew Hadlow’s attention included the “soap bubble”-like vacuolation of the neuropil, profound neuronal loss, and intense reactive astrogliosis in the absence of an associated inflammatory response. Modern neuropathologists would add only the immunohistochemical detection of abnormal forms of PrP to this list of key pathologic features of prion diseases.
Prusiner and co-workers10–12 founded the molecular era of prion biology by purifying and characterizing the infectious agent. Using a relatively rapid incubation time assay to measure hamster-adapted scrapie infectious titers, Prusiner discovered that a previously unidentified protein was the chief component of a highly infectious fraction purified from brains.13,14 In 1982, he proposed the name prion for the agent responsible for scrapie and related neurodegenerative diseases.15 The term prion initially was chosen to emphasize the hypothesis that the causative agents in these diseases were proteinaceous infectious particles that could be distinguished from viruses and viroids by their apparent lack of nucleic acid. The protein that comprised the dominant component of the infection fraction was thus named the “prion protein.”
Since the word “prion” was coined, the understanding of the biochemistry of these agents has evolved. Today, the term may refer to an alternatively folded and aggregated form of a protein that is capable of self-propagating by incorporating the normally nonaggregated host protein into the aggregate.16 A number of fungal proteins engage in prion-like behavior. Prions formed of PrP remain the only proven mammalian prions, but there is some evidence that prion-like behavior of other proteins may play a role in other neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis.17
Molecular Biology of Prion Diseases
Prion Protein
The principal component of the prion is an aggregated alternative conformation of a constitutively expressed protein. The normal form of this protein is usually designated PrPC. This is a cell surface glycoprotein, transcribed off a chromosomal gene (PRNP, located at 20p13 in humans).18,19 The protein is modified during biosynthesis by the addition of a glycosylphosphatidylinositol (GPI) moiety that anchors the carboxyl terminus to the external surface of the cell membrane.20 It may be further modified by two asparagine-linked oligosaccharides.21 Forms of PrP with no, one, or two glycosylated asparagines are produced so that PrP usually resolves into three distinct bands on Western immunoblots. PrPC is constitutively expressed at high levels in the brain, with comparable messenger RNA (mRNA) levels in neurons, oligodendrocytes, and astrocytes.22 The protein is also expressed at high levels in certain cells of the reticuloendothelial system, a distribution that is important in the pathogenesis of epidemic prion disease. The expression of the PrP mRNA does not differ between healthy and prion-infected animals.18,23 The normal function of the prion protein remains obscure. Among the candidate functions for PrPC are cell adhesion, laminin binding, neurite genesis, synaptic function, and a role in copper metabolism.24–31 Mice in which production of PrP is abolished by disruption of the PRNP gene show no gross abnormalities. On the other hand, the PrP sequence is highly conserved in evolution, so it presumably confers a survival benefit. Two human genes are homologous to PRNP, although they play no known role in neurodegenerative disease.32
Infectious Prions
Prion diseases are marked by the appearance of a pathologic form of PrP, typically designated PrPSc (Table 181-1). This pathologic form differs from PrPC in a number of biochemical properties. First, PrPSc is resistant to digestion proteases.18,33 Thus, the serine endoprotease proteinase K rapidly hydrolyzes PrPC, whereas the carboxyl-terminal region of PrPSc resists digestion in most forms of prion disease. Generation of protease-resistant carboxyl-terminal fragments of characteristic size continues to be the most widely used assay for the pathogenic form of the prion protein. Second, PrPC is readily soluble in mild non-ionic detergents, whereas PrPSc is not. Finally, studies using techniques such as circular dichroism and infrared spectroscopy indicate that approximately 40% of the PrPSc polypeptide backbone is in a β-sheet conformation, in contrast to PrPC, which contains only a very short stretch of β-sheet structure.34
TABLE 181-1
Properties of the Normal and Scrapie-Associated PrP Isoforms
| PROPERTY | PrPC (NORMAL ISOFORM) | PrPSc (SCRAPIE ISOFORM) |
| Encoded on PRNP gene on chromosome 20 | Yes | Yes |
| Present in normal brain | Yes | No |
| Present in scrapie-infected brain | Yes | Yes |
| Covalent modifications | GPI anchor, asparagine-linked oligosaccharides, single intramolecular disulfide bond | Probably identical to PrPC |
| Soluble in mild detergent | Yes | No |
| Effect of protease | Hydrolyzed to small peptides | Protease-resistant carboxyl-terminal portion of variable length |
| Secondary structure | ≈40% α-helical, little β-sheet | 30% β-sheet, 20% α-helix |
| Tertiary structure | Three α-helical regions, unstructured amino terminus | Not determined |
| Quaternary structure | Monomeric or few-subunit oligomer | Aggregated |
GPI, glycosylphosphatidylinositol; PrPC, constitutive prion protein; PrPSc, pathologic prion protein.
Modified from Riesner D. Biochemistry and structure of PrP(C) and PrP(Sc). Br Med Bull. 2003;66:21-33.
Taken together, these data indicate that PrPSc is composed of tightly bound aggregates of PrP. The aggregates sterically exclude proteases from attacking the carboxyl terminus of PrP in the aggregates, thus conferring protease resistance. The aggregates are large and dense, and thus insoluble. The presence of extensive β-sheet structure suggests that the peptides in the aggregates are bound together by intermolecular β-sheets. In contrast, PrPC exists as monomers or perhaps low-order multimers. High-resolution structures for PrPC have been obtained by nuclear magnetic resonance (NMR) spectroscopy and x-ray crystallography. These show PrPC to be a globular protein composed of three α-helical segments and two short β-strands that form a small antiparallel β-sheet. A curious feature of the protein is the absence of any regular structure of the purified protein at the amino terminus (amino acids 23 to 121) (Fig. 181-1).35–37 Presumably, this region assumes some more regular structure in vivo through association with other macromolecules or small molecule ligands. The structure of PrPSc has proved more difficult to determine, in part because available methods for high-resolution determination of protein structure require that the protein be soluble. Because the conformation of PrPSc appears to be central to the process of prion propagation, efforts to decipher this structure using advanced techniques are under way. As yet, these methods have led to no definite determination of the structure of PrPSc aggregates.38
Some molecule other than PrP might form a minor but important part of the infectious prion.39,40 There is evidence that linear polyanionic molecules, such as RNA, may facilitate the conversion of PrPC to PrPSc.39 Although RNA may be a component of natural prions, it is important to emphasize that such RNA carries no specific information; it appears that the linear polyanionic structure, rather than the specific base sequence, is important in conversion.
Other proteins, although not part of the infectious particle, may play a role in the conversion of PrPC to PrPSc. In the yeast prion state [PSI+] (see later for a further description of yeast prions), which is mediated by aggregates of the protein Sup35, the protein chaperone HSP104, as well as chaperones of the HSP70 and HSP40 families, can catalyze or inhibit the formation of prions.41,42 A perhaps a similar effect occurs in mice that overexpress HSP70 and manifest prolonged mouse-adapted scrapie incubation times relative to wild-type mice.43 The propagation of human prions in mice expressing human PrP is inhibited by the coexpression of mouse PrP. This has been taken as evidence that a factor, termed “protein X,” facilitates the conversion of PrPC to PrPSc in mice and that this factor binds mouse PrPC with greater avidity than human PrPC.44,45 Despite these lines of indirect evidence, no protein other than PrP has been proved to participate in mammalian prion propagation. Genome-wide surveys of human populations have suggested that genes other than PRNP may contribute to a risk for prion disease, but conclusive evidence is lacking.46–48
Prion diseases are endemic in several species of ruminants to which humans are exposed through food consumption and other routes. The transmission of prion diseases across species is generally less efficient than transmission within the species, a phenomenon known as the “species barrier.” One crucial determinant of the species barrier is differences between species in the amino-acid sequence of PrP. Studies in transgenic mice demonstrate that the species barrier can be abrogated by the introduction of a gene directing expression of PrP of the prion donor species into the host.49,50 Thus, mice, which are usually not susceptible to human sporadic CJD, are readily infected if the mouse Prnp gene has been replaced by a transgene coding for the human PrP sequence. This phenomenon has been exploited to generate transgenic mice that can be used as models and bioassay of bovine, ovine, human, or cervid forms of prion disease.49 A second major determinant of the efficiency of cross-species transmission of prions is the prion strain involved, as discussed later.
One of the most enigmatic features of prion diseases is the existence of what are termed prion “strains.”51 These are infectious isolates distinguishable by clinical, pathologic, and biochemical features, even in genetically identical hosts (Table 181-2). It appears that differences in the clinical manifestation of certain human prion diseases (e.g., sporadic CJD, fatal insomnia, variant CJD, and so-called “variable protease-sensitive prionopathy”) may relate to differences in the strain of the prion involved. Because the prion consists only of host-derived PrP and perhaps other host-derived components, the means by which the properties of individual strains are maintained is puzzling.
TABLE 181-2
Comparison of Two Hamster Prion Strains*
| STRAIN | ||
| PROPERTY | Hyper | Drowsy |
| Incubation time285 | 65 days | 165 days |
| Clinical signs285 | Hyperexcitability and ataxia | Lethargy |
| Size of protease-resistant fragment (nonglycosylated band)286 | 21 kDa | 20 kDa |
| Sensitivity of resistant carboxyl-terminal fragment to prolonged exposure to protease286 | Present after 24 hr of digestion | Hydrolyzed after 12 hr |
| Resistance to denaturation (concentration of guanidine HCl that denatures 50% of PrPSc)287 | 1.5 M | 1.1 M |
| Distribution of PrPSc in the brain of clinically affected hamsters288 | Most in medial geniculate nucleus and deep cerebellar nuclei | Most in regions of hippocampus, cerebellar granular layer, and occipital cortex |
| Distribution of PrPSc outside the CNS289 | In spleen and other lymphoreticular organs | Not found in lymphoreticular system |
| Species barrier285 | Nonpathogenic in mink | Pathogenic in mink |
* Perhaps the two most well-studied prion strains are “hyper” and “drowsy,” which are adapted to hamsters from transmissible mink encephalopathy, a prion disease of mink. The two strains are typically propagated in Syrian golden hamsters. Some characteristic properties are compared in the table. The number of potential strains that can exist on a single genetic background is not known, but evidence indicates that it is more than two.287
CNS, central nervous system; HCl, hydrochloride; PrPSc, pathologic prion protein.
Prion strains can be distinguished by clinical features such as incubation time or clinical signs at disease onset in genetically defined hosts.52 Strains can differ pathologically by the region of the brain in which PrPSc accumulates, by the microscopic pattern of PrPSc aggregates, by the degree of brain vacuolization, and by the morphology and distribution of PrP amyloid accumulation. Of particular importance from the point of view of human health in the presence of prion zoonoses, the barrier to transmission of prion disease between species seen with one strain of prions may be readily overcome by a different strain.51
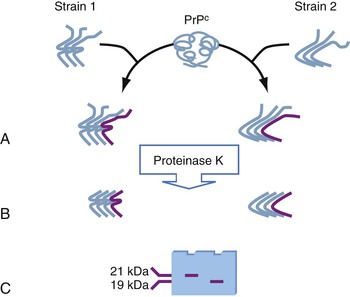
The biochemical basis of prion strain variety is understood incompletely, but it appears to reflect conformational differences between the PrPSc associated with the various strains (Fig. 181-2). The size of the protease-resistant carboxyl-terminal fragment can differ slightly between strains, as can the stability of the PrP aggregate to denaturation with chaotropic salts. Taken together, these and other observations indicate that subtle conformational differences between PrPSc associated with the various strains somehow dictate the clinical and pathologic differences.
Only a small proportion of cases of human prion disease are infectiously transmitted, but intraspecies transmission through the oral route is the major cause of prion disease in animals. The reticuloendothelial system plays a major role in the initial propagation of prions outside the central nervous system (CNS) and in carrying infection to the CNS. After oral challenge with prions, titers rise first in gut-associated lymphoid tissue. Mice deficient in the number of functional Peyer’s patches show increased resistance to oral prion challenge.53 Similarly, a number of studies document that follicular dendritic cells are necessary for mice inoculated intraperitoneally with mouse-adapted scrapie to propagate prions in the brain.54 Prion infection is carried to the brain from lymphoid tissue by axoplasmic transport in neurons of the sympathetic nervous system.55–60 In some models of scrapie infection, the vagus nerve is also important to spread infection to the CNS. The “drowsy” strain of hamster scrapie spreads to the brain without infecting lymphoreticular tissue after inoculation in the tongue, apparently by spreading through the motor neurons of the hypoglossal nerve and the gustatory sensory neurons of the nucleus of the solitary tract.61
The precise mechanism of prion propagation is not completely understood. A popular model views prion propagation as similar to the “seeded polymerization” mechanism of amyloid formation. The term amyloid is defined histopathologically as an accumulation of protein that binds the dye Congo red and alters its staining properties so that it exhibits an apple-green color under certain angles of polarized light. This occurs because amyloid is formed of fibrils composed of protein monomers, bound together in β-sheets, oriented perpendicularly to the long axis of the fibril. The kinetics of amyloid fibril formation in vitro suggests a mechanism for prion propagation: Typically, a pure solution of monomers of an amyloidogenic peptide will exhibit a lag phase, often days in duration, before the rapid conversion of monomers into amyloid fibrils. If a small “seed” fibril is introduced into a solution of monomeric peptides, the lag phase is eliminated, and the monomers rapidly polymerize. PrP amyloid of the histopathologically defined type (amyloid plaques) is seen in some forms of human and animal prion disease. Uncertainty persists over whether amyloid fibrils or a protofibrillary state may be the actual infectious moiety of prion diseases.62–64 Models of prion propagation other than seeded polymerization exist.62,65
The mechanism by which propagation of PrPSc leads to neurodegeneration remains unknown. Although the phenotypic expression of yeast prions, discussed later, is in several instances due to the loss of the protein’s normal function, this does not appear to be the case with mammalian prions. Rather, the conversion of PrPC to PrPSc causes a toxic “gain of function.”66 The nature of this toxic function remains poorly understood. Toxicity requires expression of PrP,66 and this PrP must be GPI anchored.67 As with other neurodegenerative diseases associated with protein aggregates, dysfunction of the ubiquitin-proteasome system has been invoked as a cause of neurodegeneration in prion disease.68
Yeast Prions
Wickner69 first proposed that certain epigenetic traits of yeast could be manifestations of a process analogous to that which occurs in mammalian prion diseases. To date, at least 10 yeast prions and 1 prion of the filamentous fungus Podospora anserina ([Het-s]) have been identified.70 These traits are transmitted through exchange of cytoplasm but are not linked to the mitochondrial genome.71–73 Each particular prion trait is linked to a different protein, encoded by the nuclear genome, that is found to be in an aggregated state when the prion trait is expressed.74 Unlike the mammalian PrP prion, the phenotype of at least some yeast prions is equivalent to an inactivating mutation in the cognate protein. The [Het-s] prion is particularly interesting in that it appears to convey a useful property, mating incompatibility, upon its host. Yeast prions are intensively studied as models of mammalian prions and the processes of protein folding and aggregation in general. Yeast prions do not play any role in the initiation or transmission of mammalian prion diseases.
Human Prion Diseases
Traditionally, human prion diseases have been classified by a combination of epidemiology, clinical features, histopathology, and family history. This has led to a proliferation of named syndromes that fundamentally share a similar pathophysiology. A more useful and consistent classification considers the various manifestations of human prion disease in terms of origin: sporadic, genetic, or infectiously transmitted. In general, the term Creutzfeldt-Jakob disease refers to human prion disease and includes sporadic, genetic, and infectiously acquired forms that have not been given another name. The substantial clinical and pathologic diversity of human prion disease may be the manifestations of different strains of prions. However, typing of human prion strains is an emerging technology, and the basis of prion strain differences is incompletely understood. It is not clear whether the diverse manifestations of human prion disease can be attributed to differences in the properties of the initiating prion strains or result from other as yet unidentified factors. Human genetic diversity and the variety of circumstances in which human prion disease develops or is acquired are greater than in laboratory models of prion disease, and these factors likely contribute to variance in the clinical and pathologic presentation.
The term Creutzfeldt-Jakob disease was first used by Spillmeyer in 1922 to refer to a puzzling, rapidly progressive neurodegenerative syndrome initially described separately by the eponymous German neurologists.75–77 CJD is rare, with an annual prevalence and incidence of approximately 1 case per 1 million people worldwide. In several countries, the reported rate of CJD has increased since the mid-1990s. This most likely is the result of improved surveillance in response to the outbreak of BSE and vCJD.78–82 In the United States, studies find a substantially lower rate of CJD among African Americans and other nonwhites than among whites.83,84 Whether this reflects reduced ascertainment in these groups or a truly reduced incidence is not certain.
Sporadic Creutzfeldt-Jakob Disease
Sporadic CJD (sCJD) comprises approximately 85% to 94% of all cases of human prion disease.85,86 It shows no gender predilection. Mean age at onset is 57 to 66 years, although patients as young as 17 years and older than 80 years with sCJD have been reported.86 Several studies found a peak incidence, approaching 6 per 1 million, in the eighth decade and then a distinct decline in incidence in those older than 80 years.84–86
The most distinctive clinical feature of sCJD is the pace of its progression, typically described as “rapid” or “subacute.” In the context of neurodegenerative conditions, these terms refer to perceptible declines in cognitive and motor function that are obvious over a period of a few weeks. In contrast, in more common neurodegenerative conditions, such as Alzheimer’s or Parkinson’s disease, decline is typically only apparent over periods ranging from months to years. Some observers have noted that the pace of decline in CJD accelerates until the later stages of akinetic mutism, when neurologic dysfunction is so severe that it is difficult to appreciate further decline. A second distinctive feature of CJD is the prominent involvement of multiple brain systems in which motor signs, such as ataxia, bradykinesia, or spasticity, are combined with memory and other cognitive deficits. There is a great deal of variability in the clinical manifestations of CJD, and this has led to attempts to describe a variety of clinical subtypes, including those with predominance of visual,87 cerebellar,88 thalamic,89 and striatal90,91 features. The existence of these syndromes indicates that CJD may affect particular brain regions disproportionately. In some cases, the particular regional predominance of pathology may reflect the strain of prion involved.
In many patients with sCJD, there is a prodromal phase of psychiatric disturbance before the onset of neurologic signs. Of 126 mostly sporadic cases of CJD, 26% of patients had psychiatric signs in the prodromal or presenting phase.92 Most common were sleep disturbance, depression, and anxiety. In approximately one third of patients, prominent initial visual or cerebellar symptoms may overshadow dementia. Mental deterioration typically is rapidly progressive, and the average duration of illness from onset of symptoms to death is 7 to 9 months. In addition to profound and rapidly progressive mental deterioration, another very common feature is myoclonus. However, myoclonus in demented patients is not pathognomonic of CJD. It can occasionally occur in Alzheimer’s disease and is common in Lewy body dementia. Extrapyramidal and cerebellar signs, including bradykinesia, rigidity, ataxia, nystagmus, and tremor, ultimately develop in approximately two thirds of patients. Approximately 40% to 80% of patients have signs of corticospinal tract dysfunction, including hyperreflexia, spasticity, and extensor plantar responses. Prominent visual disturbances, which can include visual field cuts, cortical blindness, and visual agnosia, occur in 50% of CJD patients.
Some patients have vague sensory complaints, including pruritus and aching limbs. It is unclear if these sensations are of peripheral or central origin.93 Signs of a motor or sensory peripheral neuropathy are occasionally found in sCJD, although these signs are almost always overshadowed by the dramatic signs of CNS dysfunction. One study found clinical evidence of peripheral neuropathy in approximately 20% of cases examined and electrophysiologic abnormalities in 14 of 16 surveyed cases of sCJD.94 On occasion, signs of sensory or motor neuropathy may dominate the early disease course. Rarely, fasciculations and muscle wasting will be so prominent as to suggest amyotrophic lateral sclerosis.95,96 Some cases of CJD also have been reported in which the clinical features indicated prominent autonomic nervous system involvement. These features included hypohidrosis, bowel dysfunction, abnormal pupillary responses to autonomic drugs, abnormal diurnal blood pressure variation, and electrocardiogram abnormalities. Such findings are a cardinal feature of FFI.
Certain neurologic disturbances occur only rarely as prominent features in CJD, and their presence should prompt clinicians to consider other diagnostic possibilities. Although seizures occur in 10% to 20% of cases, they are rarely a dominant feature and typically are amenable to therapy. Cranial nerve involvement is never prominent, although isolated cases have been reported with involvement of the pupils; extraocular movements; and trigeminal, auditory, and vestibular systems.97
At least two distinctive sporadic forms of CJD bear the hallmarks of discrete prion strains. First, very rare sporadic cases of a clinical and pathologic syndrome indistinguishable from FFI (see “Genetic Creutzfeldt-Jakob Diseases”) will be encountered in patients without PRNP mutations.98,99 This form of sporadic prion disease maintains a biochemical signature identical to that of FFI upon serial transmission in transgenic mice expressing human PrP. It thus behaves as a distinct human prion strain.
Second, a recently described condition, “variable protease-sensitive prionopathy” is a form of sporadic CJD with fewer and less prominent motor and sensory signs and a pattern of cognitive impairment said to resemble frontotemporal dementia (although a few published clinical descriptions suggest the pattern of dementia may sometimes be similar to Alzheimer’s disease).100,101 Progression is slower than typical sCJD, with a mean duration of 30 months. PrPSc from these patients is more sensitive to protease digestion than in typical sCJD. The predominant protease-resistant fragment is 8-kDa and cleaved at both the amino and carboxyl termini, whereas in sCJD, the predominant fragment is cleaved only at the amino terminus and is 19- or 21-kDa in size. The disease is quite rare; 13 cases were identified by the main U.S. prion disease pathology center over an 8-year period, and 5 cases were identified in Britain in a retrospective review of 20 years.101,102 Ignoring ascertainment problems, which may be significant, these translate into an annual incidence of ≈0.5 per 100 million in either population. The disease has been successfully transmitted (to an unusual assay animal, the bank vole), although the serial transmissions needed to confirm it as a distinct strain are still in progress.101,103
Genetic Creutzfeldt-Jakob Diseases
Approximately 10% of cases of human prion disease are caused by some nonconservative (i.e., amino-acid sequence–changing) mutation in PRNP, although the precise proportion varies among countries.85 PRNP mutations are found in all cases from families with a history of inherited prion disease, as well as approximately 5% of apparently sporadic cases. Familial prion diseases are transmitted in an autosomal-dominant pattern, usually with high, but not always complete, penetrance.
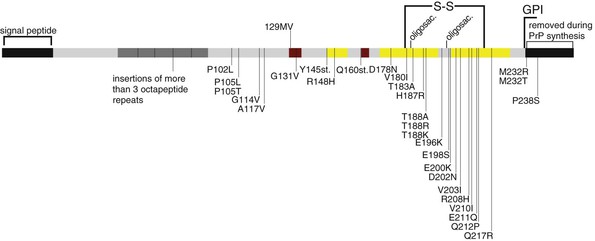
At least 30 distinct mutations in the PRNP gene are known to be associated with the inherited prion diseases (see Fig. 181-1).104,105 Not all patients with genetic forms of prion disease present with a family history of dementia.106 In a large European survey of prion disease, a family history of a CJD-like dementia was elicited in only half of genetic cases.107
Specific mutations in the PRNP gene tend to be associated with particular clinical and pathologic disease phenotypes.107 However, significant variability is seen both within and between kindreds harboring identical mutations. The disease associated with many mutations is essentially indistinguishable from sporadic CJD. Among these is the most common mutation worldwide, a lysine-for-glutamic acid substitution in codon 200 (E200K).105 This mutation has been found in geographic clusters of familial CJD in Slovakia and Chile and among Sephardic Jews in Greece, Libya, Tunisia, and Israel. Before the mutation was discovered to be prevalent in Libyan Jews, the high rate of CJD in this group was misattributed to the consumption of sheep brains or eyeballs.108 The median age of onset is 58 years. The mutation is highly but incompletely penetrant so that approximately 80% of carriers will develop CJD by the eighth decade of life.109 Because the mutation is found in inbred populations, rare carriers are homozygous for the mutation. Surprisingly, these people seem to develop illness only slightly earlier than heterozygotes (50 vs. 59 years).110
Some PRNP mutations are associated with syndromes markedly different from sCJD.
Gerstmann-Straüssler-Scheinker Syndrome
In some families with inherited prion disease, most victims develop prominent early ataxia and signs of corticospinal tract degeneration. This picture is often associated with accumulations of PrP in large amyloid plaques in the CNS.111,112 This clinicopathologic picture is known as Gerstmann-Straüssler-Scheinker syndrome (GSS). The syndrome is linked to several different mutations in PRNP, including P102L, P105L, A117V, Q187H, F198S, D202N, Q212P, Q217R, Y145STOP, and an insertion of eight or nine octapeptide repeats.113 The duration of illness ranges from 3 months to 13 years, with a mean of 5 or 6 years.113,114 Overt dementia occurs late in the disease.
Neuropathologic findings of GSS are typical of other prion diseases, except that patients have widespread amyloid plaques composed of densely aggregated PrP. Within the cerebellum, where the concentration is typically the most dense, plaques are found in the molecular layer, are often multicentric, and are associated with a microglial reaction. The multicentric morphology of GSS plaques distinguishes them from plaques seen in kuru, which are typically unicentric.115 The degree of spongiform change is variable, ranging from substantial and severe to completely absent. In cases in which plaque pathology predominates and spongiform pathology is absent, Western blot analysis of protease-resistant PrP finds mainly an unusual 8-kDa fragment, as opposed to the 21-kDa or 19-kDa fragment associated with most forms of human prion disease. Like many genetic forms of prion disease, GSS is transmissible to transgenic mice expressing the human form of PrP. Cases with almost all plaque-type pathology transmit a plaque-predominant disease to these mice, whereas those with significant spongiform-type pathology transmit a more typical spongiform disease.116 Thus, two seemingly different prion strains can propagate in persons with GSS.
Fatal Familial Insomnia
FFI first was reported as a human disease in 1986,117 although there is clinical and pathologic overlap between FFI and cases described earlier as “thalamic dementia.”118 In the early 1990s, the immunohistochemical detection of abnormal PrP and the subsequent recognition of a D178N mutation in PRNP marked FFI as a prion disease with unusual features.119 The D178N mutation can occur in two different PRNP allele forms, with either a methionine or a valine code for the polymorphic codon 129. The D178N/129M allele associates with FFI, whereas a syndrome more closely resembling typical sCJD occurs in patients carrying the D178N/129V allele. Although the first reports of FFI were all in Italian families, currently, at least 27 families with FFI have been reported from the United Kingdom, Europe, the United States, Finland, Australia, China, and Japan.120
In the typical form of FFI, patients develop insomnia as a prominent and early complaint, along with signs of autonomic hyperactivity (increased sweating, tearing, salivation, mild nocturnal hyperthermia, tachycardia, and hypertension). Motor disturbances develop later and can include ataxia, myoclonus, spasticity, hyperreflexia, and dysarthria.119,120,121 Marked memory impairment is not prominent early in the disease, although a delirium-like hallucinosis may occur. The mean age of onset in FFI is 50 to 56 years, somewhat younger than in sCJD, but cases in patients as young as 19 years and as old as 83 years of age have been reported.107,122–124 Series of patients with the FFI genotype describe substantial clinical heterogeneity. Ataxia and psychiatric signs, such as depression, apathy, or anxiety, are common initial complaints, and insomnia is not always noted by the patient, family, or clinicians.122,123 Polysomnography is a sensitive test for FFI, being abnormal in almost all cases, but investigations have shown this is true for sCJD as well.125 In both forms of prion diseases, there are marked reductions or absence of both rapid eye movement (REM) and normal non-REM sleep.
Neuropathologic changes, including neuronal loss and reactive gliosis, are found consistently in the anterior ventral and mediodorsal nuclei of the thalamus, the inferior olives, with less prominent involvement of the cerebellar and cerebral cortex.114,121,126 Immunostaining of brain material for PrPSc is positive, although the concentration of protein is 5 to 10 times less than that seen in sCJD.
As mentioned earlier, a rare sporadic form of human prion disease presents with clinical and histopathologic features identical to those of FFI (sporadic fatal insomnia [sFI]).98 Both FFI and sFI can be transmitted to transgenic mice expressing wild-type human PrP. The clinical and histopathologic properties of the disease in these mice, as well as the biochemical properties of the PrPSc produced, are identical, and they are distinct from those of sCJD.99 Thus, the prions associated with sFI and FFI are a distinct strain, different from that which causes typical sCJD.
Long-Duration Disease
Certain PRNP mutations frequently cause disease with exceptionally slow progression. These include large expansions in the octapeptide repeat region and the missense mutations T183A and H187R.127–130 Some mutations may be associated with lifelong psychiatric disturbances that precede the onset of progressive dementia by decades.129
Polymorphisms in PRNP
Polymorphisms at codon 129 of PRNP play a role in CJD expression and susceptibility. In European populations, approximately 60% to 70% of alleles have methionine at this position, and the rest have valine. The alleles are in Hardy-Weinberg equilibrium, so genotypes in the general population are approximately 42% 129 MM, 46% MV, and 12% VV.131 In contrast, 95% of patients who develop sCJD exhibit homozygosity (either MM or VV) at this locus.132,133 Codon 129 allele distributions vary across ethnic groups, but the tendency for overrepresentation of homogzygotes in CJD cases holds.134 Genome-wide association surveys have demonstrated that the codon 129 polymorphism genotype is a significant risk factor for both sporadic and acquired CJD. Weaker associations with other loci within PRNP and outside of this gene have been inconsistently reported and are of uncertain significance.46–48
PRNP polymorphisms might influence the risk of neurodegenerative conditions other than CJD. Controversial and inconclusive epidemiologic evidence suggests that the PRNP polymorphism at codon 129 may influence the risk of developing Alzheimer’s disease.135–137 These observations are given some support by studies showing that PrPC binds oligomers of Aβ, the protein that aggregates to cause Alzheimer’s disease, at neuronal synapses.138
Infectiously Transmitted Human Prion Disease
In most countries, less than 1% of human prion disease is infectiously acquired. Yet, study of an epidemic of infectious prion disease in New Guinea led to the recognition of prions as a cause of neurodegenerative illness in humans and transmission of prion disease from animals or humans to humans continues to be an issue of great concern.
Kuru
Kuru is a neurodegenerative disease that was endemic within the Fore linguistic tribal group of the Eastern Highlands of Papua New Guinea.6,139 It is thought to have arisen in the early 20th century and have been transmitted through the practice of ritual endocannibalism at funeral feasts.140 No one born since the cessation of this practice has developed kuru.140 The mean incubation period was 10 to 13 years, with 90% of cases occurring within 21 to 27 years of exposure. The incubation period was likely related to exposure dose and was shorter in women than in men and shorter in older women than in younger women, reflecting the likelihood of participation in cannibalistic practices.141 Rare cases of kuru continue to occur, likely as a result of exposure before ritual cannibalism ceased in 1960. Thus, in some cases, the incubation period for kuru can exceed 50 years.140
Kuru remains important conceptually, even as it disappears as a clinical entity, as the largest outbreak of human-to-human transmission of a prion disease and as an example of human prion disease transmitted via the oral route. In its clinical and pathologic manifestations, it is distinct from most human prion disease. Kuru typically begins insidiously with a prodrome of headaches and aching limbs that may last for several months.142 This prodrome is followed by the development of an inexorably progressive neurologic disease resulting in death within 3 to 24 months of onset, usually from intercurrent pneumonia and malnutrition. Typical disease duration is 12 months.142 The cardinal clinical features include cerebellar ataxia, action tremor, and involuntary movements (choreoathetosis, myoclonic jerks, and coarse fasciculations). Dementia is usually not noted until late in the illness, 8 or more months after the onset of ataxia.
Neuropathologic examination of kuru brains shows neuronal loss, astrogliosis, and the accumulation of PrPSc, all findings typical of prion disease.143–146 The pathologic hallmark of kuru is the presence of PrP amyloid plaques, predominantly in cerebellar tissue. These plaques are usually unicentric, located in the granular layer of the cerebellum, and often associated with microglial cells. Kuru cases show a higher-than-expected incidence of MM homozygosity at polymorphic codon 129 of the PRNP gene, as is seen in sCJD. In contrast, older women who potentially were exposed to infected brain material during the era of cannibalism and survived without developing kuru show a higher-than-expected frequency of heterozygosity at codon 129. Thus, this heterozygosity may have played a protective role against the transmission of prion diseases among the Fore. More strikingly, a PRNP G127V mutation is found only among survivors of the kuru epidemic and their descendants, and in no other population in the world.147 This mutation appears to have conferred resistance to kuru and to have been strongly selected for during the epidemic.
The properties of kuru prions on transmission to transgenic mice expressing human PrP suggest that it is the same strain as that most commonly found in sporadic CJD.148 This finding is consistent with the hypothesis that the kuru epidemic arose out of a naturally occurring case of sCJD that was then spread by cannibalism. It is puzzling that the clinical and pathologic features of kuru are quite distinct from those of typical sCJD. It is possible that the oral route of exposure alters the clinical and pathologic presentation, or that current methods of typing strains are insensitive to differences between kuru and sCJD.
Variant Creutzfeldt-Jakob Disease
Beginning in 1995, cases of a new variant of CJD were reported from the United Kingdom.149,150,151 As of April 2013, a total of 176 cases have been reported to the U.K. Creutzfeldt-Jakob Disease Surveillance Unit (see www.cjd.ed.ac.uk/ for latest case totals). This includes 3 symptomatic “secondary” cases transmitted through nonleukocyte-depleted red blood cell transfusion and 2 asymptomatic cases—1 from red blood cell transfusion and the other probably transmitted through pooled plasma-derived factor VIII.152–154 An additional 51 cases have occurred outside the United Kingdom, with 27 of these in France and the remainder in the Republic of Ireland, Italy, the Netherlands, Portugal, Spain, Japan, Saudi Arabia, the United States, and Canada.155 Of the 5 North American cases, 4 appear to be due to exposure of the victims to infected beef products in the United Kingdom or Saudi Arabia.155,156 Retrospective review of available autopsy material suggests that vCJD did emerge as a new disease entity in 1995; no current evidence suggests that cases occurred earlier.157,158 It is likely that millions of people were exposed to BSE-infected food.159 However, fears of a large epidemic of vCJD have not been realized. The largest number of annual cases, 28, was reported in 2000.155 The incidence of vCJD is declining; there were no deaths from vCJD in Great Britain in 2012 and only 2 other cases in the world, both in France.155
The epidemiologic, clinical, and pathologic features of the vCJD cases set them apart from typical sCJD.150,160,161 Patients with vCJD have been considerably younger than patients with sCJD, with a mean age at onset of 26 years (range, 12 to 74 years) compared with 65 years for sCJD. Ninety percent of vCJD patients are younger than 40 years at the onset of their disease. The reason for this striking age dependence is not clear, but it appears to be due to greater susceptibility in the young rather than greater exposure to BSE-contaminated foodstuffs.162 The duration of illness in vCJD is longer (average, 14 months) than in sCJD (average, 4.5 months). This longer duration may be a reflection of the younger age of the patients because younger patients with sporadic CJD have a similar duration of illness.163,164
Patients with vCJD frequently present with sensory disturbances and psychiatric manifestations. Such symptoms are not unusual in young-onset sCJD, but in vCJD, they are more prominent, perhaps because dementia and more obvious neurologic signs supervene later than in sCJD.163 Among the sensory symptoms are vague pain, cold sensation, or paresthesias involving the face and limbs or in a hemisensory distribution. Most studied patients have had normal electromyography or mild abnormalities that do not point to a peripheral origin for the sensory disturbance.93 It is speculated that they may be of thalamic origin.165 Psychiatric manifestations frequently include dysphoria, withdrawal, anxiety, irritability, insomnia, and loss of interest in usual activities.166,167 These symptoms often prompt an initial diagnosis of psychiatric illness. Neurologic signs and symptoms are uncommon within 4 months of onset. As disease progresses, the most frequent neurologic signs include dysarthria and gait disturbance. In the later stages of disease (>6 months after onset), prominent neurologic signs include hyperreflexia, myoclonus, incoordination, or other cerebellar signs.166
Blood products have very likely transmitted vCJD on at least five occasions. Among a group of 66 individuals identified as having received blood components from donors who later developed vCJD, only 33 of the transfusion recipients survived more than 5 years after transfusion, with the rest dying of conditions other than CJD or dementia.153,168–170 Three of the remaining 33 recipients developed vCJD 6 to 8 years after transfusion. The donors had donated 16 months to 3.5 years before the onset of vCJD. One donor was linked to vCJD cases in 2 recipients. All cases received nonleukocyte-depleted red blood cells. The rate of vCJD in transfusion recipients from donors, who later developed vCJD, 4 per 66 or 6%, is four orders of magnitude greater than the rate of vCJD in the British population, conclusively implicating the transfusion as the cause of disease. Remarkably, in an autopsied case, PrPSc was found in the tonsils, suggesting that the accumulation of PrPSc within tonsils that is found in most vCJD cases is not due to the alimentary route of exposure but, rather, is a tropism inherent in the vCJD prion strain.152 Another of these 33 recipients died of causes unrelated to vCJD but was found at autopsy to have PrPSc in the spleen.171 Finally, a survey of tissue obtained at autopsy from 11 persons with hemophilia, known to have received U.K.-sourced, pooled, plasma-derived clotting factor concentrates, found PrPSc in a single spleen.172 Both cases of asymptomatic infection occurred in blood product recipients who were heterozygous (M/V) at PRNP codon 129, whereas that codon was homozygous M/M in the 3 symptomatic cases.
The neuropathologic features of vCJD are strikingly different than those of sCJD.150 The most characteristic differences between the neuropathology of vCJD and sCJD seem to be the prominent involvement of the cerebellum in almost all cases of vCJD, compared with only a subset of cases with sCJD (Brownell-Oppenheimer variant and GSS cases). vCJD cases also show typical spongiform change, neuronal loss, and astrogliosis in the cortex, basal ganglia, and thalamus, but these do not distinguish vCJD from sCJD. vCJD cases have prominent amyloid plaques distributed throughout the cerebrum and cerebellum and, to a lesser extent, the basal ganglia and thalamus. These plaques have a dense eosinophilic center and pale periphery and are surrounded by vacuoles in the neuropil arranged around the plaque like petals on a flower (hence, the name “florid plaques”). The plaques stain strongly positive for PrPSc. This flower-like morphology is common in plaques from vCJD but is infrequently seen in other plaque-forming types of human prion disease, such as GSS, kuru, and some sporadic cases of CJD.173 Patients with vCJD also have a consistent pattern of electrophoretic mobility of PrPSc protein (type 4 isoform) that is distinct from the mobility patterns encountered in sCJD and is similar to the pattern seen in BSE PrPSc.174,175,176
Compelling evidence indicates that vCJD is the result of bovine-to-human transmission of BSE.175,177–180 From an epidemiologic standpoint, cases of vCJD followed a massive epidemic of BSE in the United Kingdom, with a lag period that is consistent with the known incubation period of prions. During the BSE epidemic, among the first cases, which were recognized retrospectively as early as April 1985, it was estimated that several hundred thousand BSE-infected cattle might have entered the human food chain.181–183 The number of BSE-infected cattle peaked during 1992 and 1993 and subsequently declined steadily. This decline has been attributed to bans on using ruminant protein for ruminant feeds (July 1988) and on using bovine brain, spinal cord, and other specified offal as feed for nonruminant animals and poultry (September 1990). Another ban prohibited use of certain bovine tissues for human consumption (November 1989). It has been suggested that the BSE epidemic was triggered by changes in the rendering process, particularly the abandonment of the use of organic solvents.184
Additional evidence for a BSE-vCJD link comes from the close neuropathologic similarities between the two diseases. Transgenic mice expressing bovine PrP develop indistinguishable neurologic illness and neuropathologic changes after a similar incubation period when injected with brain material from either cattle with BSE or humans with vCJD, and this differs from the pattern and incubation time seen after inoculation with scrapie.180 In addition, the PrPSc protein isoforms isolated from the brains of the BSE-inoculated or vCJD-inoculated mice show an identical fragment size and glycosylation pattern, which differs from that seen after scrapie inoculation.179 Furthermore, macaques inoculated with BSE prions develop florid plaques histologically indistinguishable from those seen in vCJD, whereas those inoculated with sporadic CJD prions do not develop plaques.178 Finally, the susceptibility of mice expressing murine PrP, or transgenic mice expressing PrP of humans, to vCJD more closely resembles the susceptibility of these same mice to BSE than to other forms of human prion disease.185
With one possible exception, all vCJD patients tested to date are homozygous for methionine at polymorphic codon 129 (129M).151,174 One young patient in Great Britain with the 129VV genotype developed CJD with clinical and pathologic features not typical of sporadic or variant CJD. Protease-resistant PrPSc from this patient showed a size and glycoform pattern identical to that found in vCJD.186 Whether this was a sporadic case or, like vCJD, caused by exposure to BSE prions is undetermined. It is fortunate, though puzzling, that despite the widespread exposure of populations to BSE, comparatively few persons have developed vCJD. As mentioned earlier, genome-wide association studies have sought loci that might confer exceptional susceptibility to vCJD upon some people. No convincing evidence for genetic risk factors, except for homozygosity for methionine at PRNP codon 129 has been found.46,47
The wide exposure of the British population to BSE-contaminated food products and the sometimes very long incubation period of prion diseases have raised concerns that human infection with BSE prions may be more prevalent than is apparent from the number of clinical cases. In this regard, in certain animal models of transmission of prions across species, animals can be efficiently infected, as indicated by demonstrable prion replication in the spleen and other organs of the reticuloendothelial system, but never show clinical signs of disease. Immunohistochemical surveys of large numbers of archived tonsils and appendices has been undertaken in Great Britain to look for subclinical prion infection.187–190 A survey of approximately 11,000 appendices collected between 1995 and 1999 found 3 with immunohistochemically detected PrPSc.187 The study was repeated on approximately 32,000 appendices collected between 2010 and 2012, and 16 were found to harbor PrPSc.190 Together, these studies suggest that about 1 in 2000 Britons may be subclinically infected with prions. However, as yet, no study has been performed on a control group, for instance, a population not known to have exposure to BSE. Therefore, it is uncertain whether these findings are related to BSE exposure, represent some other aspect of prion biology, or are artifactual. If the PrPSc detected in appendices is the result of BSE prions propagating in human lymphoid tissue (i.e., subclinical vCJD), then it is possible that a large number of symptomatic cases could eventually occur and that donated blood products, organs, or tissues from subclinically infected Britons might transmit prion disease to recipients.
Diagnosis of vCJD should be considered when patients with a history of residence in a BSE endemic area, such as the United Kingdom, develop a progressive neurodegenerative disease. Table 181-3 lists the World Health Organization criteria for vCJD. These criteria identified vCJD cases with 77% sensitivity and 100% specificity.161
TABLE 181-3
Case Definitions for Sporadic and Variant Creutzfeldt-Jakob Disease
| Sporadic CJD |
| Possible |
EEG atypical or not known, and At least two of the following four clinical features: Visual or cerebellar disturbance Pyramidal/extrapyramidal dysfunction Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





