Principles of pediatric oncology
Teena Bhatla, MD  William L. Carroll, MD
William L. Carroll, MD
Overview
Pediatric malignancies differ from those that occur in adults in their relative incidence, pathological type, clinical presentation, and prognosis. The last several decades of the twentieth century have been a period of enormous accomplishment in the field of pediatric cancer research and outcomes have steadily improved largely due to results from large well-controlled clinical trials conducted by various cooperative groups at the national and international level. This chapter seeks to provide a broad overview of pediatric oncology, outlining the several common cancer types that are unique to the pediatric age group.
Introduction and epidemiology
Cancer is a relatively rare disease in childhood, yet cancer remains the second leading cause of death in children after accidents. The spectrum of cancer types in children is distinctly different compared to adults. Although epithelial tumors (carcinomas) dominate the adult cancer spectrum, childhood cancers tend to be of hematopoietic (e.g., leukemias, lymphomas), mesenchymal (sarcomas), and neuroectodermal (e.g., neuroblastomas (NBs), gliomas, medulloblastomas) origin. Acute lymphoblastic leukemia (ALL) and brain tumors are the most common childhood cancers accounting for 25% and 20%, respectively, of all cases. The incidence of the most common childhood tumors is illustrated in Figure 1 and Table 1, but it should be noted that incidence can vary according to ethnic and geographic factors. For example, Burkitt lymphoma accounts for 50% of all childhood cancer in equatorial Africa.
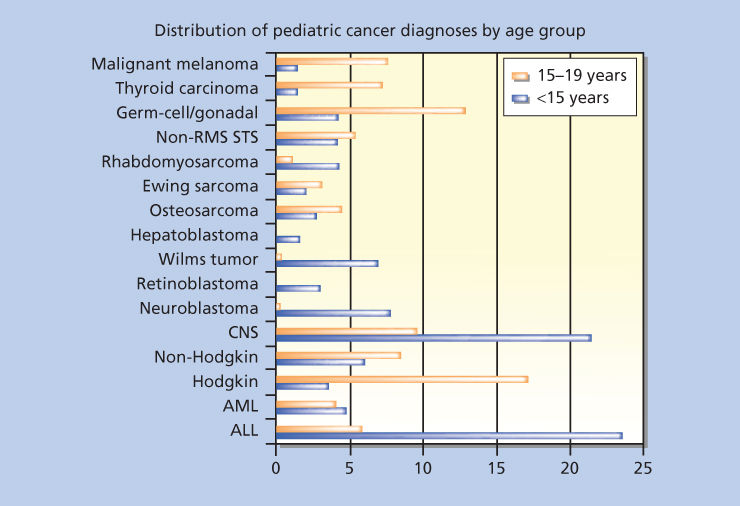
Figure 1 Distribution of pediatric cancer diagnoses by age group. Abbreviations: ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; CNS, central nervous system; RMS, rhabdomyosarcoma; STS, soft tissue sarcoma.
Table 1 Age-adjusted and age-specific SEER annual incidence rates per million population for childhood cancer, 2007–2011
| Cancer type (ICCC group) | Age at diagnosis | ||||||
| 0–14 | 0–19 | <1 | 1–4 | 5–9 | 10–14 | 15–19 | |
| All groups combined (including benign CNS tumors) | 172.8 | 191.2 | 253.4 | 228.1 | 137.2 | 150.9 | 246.2 |
| All leukemia | 54.4 | 49.4 | 51.7 | 95.0 | 44.5 | 33.9 | 34.5 |
| Acute lymphoblastic leukemia (ALL) | 41.9 | 35.9 | 20.3 | 79.7 | 37.1 | 22.1 | 18.2 |
| Acute myeloid leukemia (AML) | 8.0 | 8.5 | 18.9 | 10.6 | 4.5 | 7.4 | 9.9 |
| All CNS | 44.3 | 45.1 | 48.8 | 49.4 | 43.1 | 40.6 | 47.8 |
| Ependymoma and choroid plexus tumors | 4.0 | 3.7 | 8.8 | 6.9 | 2.6 | 2.4 | 2.8 |
| Astrocytoma | 16.7 | 15.6 | 15.5 | 18.7 | 17.1 | 15.2 | 12.2 |
| Embryonal tumors | 7.6 | 6.4 | 12.5 | 11.3 | 7.4 | 4.1 | 2.7 |
| Hodgkin lymphoma | 5.9 | 12.4 | — | 1.1 | 5.0 | 11.6 | 31.8 |
| NHL (excluding Burkitt lymphoma) | 6.8 | 8.6 | — | 4.5 | 6.9 | 9.5 | 14.1 |
| Burkitt lymphoma | 2.6 | 2.6 | — | 1.7 | 3.4 | 3.0 | 2.4 |
| Neuroblastoma and ganglioneuro blastoma | 10.6 | 8.1 | 51.0 | 20.9 | 4.3 | 1.2 | 0.9 |
| Retinoblastoma | 4.2 | 3.1 | 27.7 | 8.7 | — | — | — |
| Wilms’ tumor and other nonepithelial renal tumor | 8.4 | 6.8 | 15.3 | 19.8 | 5.8 | 1.1 | 1.9 |
| Hepatoblastoma | 2.9 | 2.5 | 10.1 | 6.4 | 0.8 | 0.9 | 1.5 |
| Osteosarcoma | 4.2 | 5.1 | — | — | 3.3 | 8.5 | 7.7 |
| Ewing sarcoma | 2.5 | 2.8 | — | 1.1 | 2.2 | 4.3 | 3.5 |
| Soft tissue sarcomas | 11.2 | 12.4 | 19.6 | 11.0 | 8.8 | 12.2 | 15.6 |
| Rhabdomyosarcoma and embryonal sarcoma | 5.4 | 4.9 | 5.8 | 7.7 | 4.9 | 4.1 | 3.5 |
| Germ cell tumors | 6.0 | 12.2 | 18.9 | 4.0 | 3.2 | 7.8 | 30.7 |
| Carcinomas | 6.8 | 17.6 | — | 1.8 | 4.0 | 14.3 | 49.7 |
| Adrenocortical carcinoma | 0.3 | 0.2 | — | — | — | — | — |
| Thyroid carcinoma | 2.8 | 8.3 | — | — | 1.3 | 6.8 | 24.6 |
| Nasopharyngeal carcinoma | 0.3 | 0.6 | — | — | — | 0.8 | 1.4 |
| Malignant melanoma | 1.8 | 4.3 | — | 0.9 | 1.3 | 3.1 | 11.7 |
There is a suggestion that the incidence of cancer is rising particularly among certain subtypes of childhood tumors, but better reporting of cases and improved diagnostic imaging may account for some of this trend.1 The etiologies of most childhood cancers remain uncertain. Environmental influences are less likely to play a role in children with radiation being the most well-established risk factor. Breakthroughs in next-generation sequencing technologies also indicate that childhood tumors have a much lower mutational burden compared to malignancies of adults, and this would also suggest that the environment plays a less important role in tumor initiation.2
Genetic predisposition causes about 5% of all cancers in the pediatric age group although this figure may increase with recent results from next-generation sequencing revealing unrecognized mutations in cancer predisposition genes. About a third of all cases of retinoblastoma (RB) are caused by a germline mutation in RB1 and children with Down syndrome (DS) have 20-fold increased incidence of leukemia (lymphoid and myeloid). Other hereditary syndromes, such as neurofibromatosis 1 (brain tumors), Beckwith–Wiedemann [Wilm’s tumor (WT), hepatoblastoma (HB), rhabdomyosarcoma (RMS)], Li–Fraumeni (sarcomas, carcinomas), Gorlin’s (medulloblastoma, skin cancer), and ataxia telangiectasia (leukemia, lymphoma), account for a small, but important fraction of childhood cancers. Often these familial syndromes paved the way for identification of genes that play a role in the much more common sporadic forms of the disease.
In addition, more subtle “host” genetic variation (compared to the syndromes described above) has been suggested by the identification of genetic variations (single nucleotide polymorphisms or “SNPs”) that account for an increased risk of cancer and may impact on outcome and side effects of therapy. For example, germline SNPs in ARID5B, IKZF1, CEBPE, PIP4K2A, and CDKN2A-CDKN2B influence susceptibility to ALL and GATA3 variants are associated with a particular subtype of ALL called “Ph-like” ALL (see below).3
The dramatic improvement in outcome for childhood cancer represents one of the greatest success stories in the history of the “War on Cancer” that began in earnest in 1971. At that time, roughly 60% of all children less than 20 years of age survived 5 years from diagnosis, whereas today, the figure is more than 80% (Figure 2 and Table 2). There are many reasons for these advances including the commitment to multidisciplinary care and highly disciplined clinical trials directed by nation and worldwide consortiums such as the Children’s Oncology Group (COG). Interestingly, many of the same chemotherapeutic agents developed decades ago are still used today, but augmentation of doses and schedules have been realized through advances in supportive care and treatment intensification has improved outcome for almost all tumors. Lastly, risk-adapted therapy, tailoring treatment based on prognostically relevant clinical and laboratory variables, has allowed intensification of treatment for those patients most likely to benefit while avoiding more toxic therapy for those patients predicted to have an excellent outcome with standard treatment. Survival rates have been most dramatic for ALL (10% in the 1960s to greater than 90% today), the most common malignancy, but survival rates remain low for patients with certain brain tumors, metastatic solid tumors, and relapsed disease. Moreover, while mortality has decreased by approximately 50%, close to 2000 children die each year in the United States.4 Thus, more effective treatments are urgently needed, and the cost of cure is substantial with short and long side effects, so more targeted, less toxic treatments are a priority also.
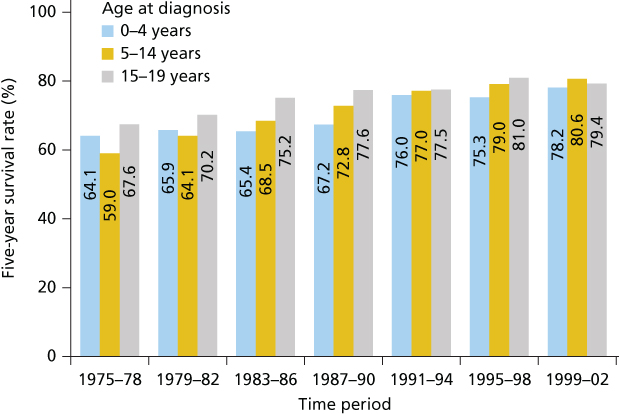
Figure 2 Five-year survival rates for all cancers by age group and period of diagnosis from 1975–2002. Source: Smith et al. 2010.5 Reprinted with permission. © 2010 American Society of Clinical Oncology. All rights reserved.
Table 2 Five-year relative survival rates (percent) (SEER.cancer.gov/csr/1975_2011)
| Site (ages 0–14) | 1975–1977 | 1978–1980 | 1981–1983 | 1984–1986 | 1987–1989 | 1990–1992 | 1993–1995 | 1996–2002 | 2004–2010 |
| All sites | 58 | 62.6 | 66.7 | 68.2 | 71.4 | 75.4 | 77.0 | 79.4 | 83.1 |
| Bone and joint | 51.3 | 48.7 | 56.8 | 59.4 | 66.8 | 66.8 | 73.9 | 71.6 | 78.9 |
| Brain and CNS | 56.9 | 57.7 | 56.0 | 61.8 | 63.8 | 64.0 | 70.1 | 74.1 | 74.1 |
| Hodgkin lymphoma | 80.3 | 87.7 | 87.7 | 90.8 | 87.1 | 96.7 | 94.6 | 95.3 | 97.6 |
| ALL | 57.6 | 66.4 | 71.3 | 72.5 | 77.7 | 83.0 | 83.8 | 87.0 | 91.8 |
| AML | 18.8 | 25.8 | 26.7 | 30.0 | 36.2 | 41.0 | 41.0 | 53.1 | 66.3 |
| Neuroblastoma | 52.4 | 56.9 | 54.5 | 52.0 | 61.9 | 76.6 | 67.2 | 68.7 | 76.6 |
| NHL | 42.6 | 52.7 | 66.9 | 70.3 | 70.7 | 75.9 | 80.6 | 86.0 | 85.6 |
| Soft tissue | 61.0 | 74.3 | 69.2 | 73.4 | 65.4 | 79.2 | 76.4 | 71.7 | 81.3 |
| Wilms tumor | 73.1 | 78.6 | 86.4 | 90.7 | 92.2 | 91.9 | 91.4 | 92.1 | 91.7 |
| Site (ages 0–19) | 1975–1977 | 1978–1980 | 1981–1983 | 1984–1986 | 1987–1989 | 1990–1992 | 1993–1995 | 1996–2002 | 2004–2010 |
| All sites | 61.5 | 65.2 | 68.0 | 70.6 | 73.3 | 76.0 | 77.4 | 79.5 | 83.5 |
| Bone and joint | 51.1 | 48.4 | 51.2 | 57.2 | 63.5 | 68.2 | 68.6 | 68.0 | 75.5 |
| Brain and CNS | 58.7 | 58.1 | 57.6 | 64.0 | 65.9 | 66.1 | 71.0 | 75.1 | 74.0 |
| Hodgkin lymphoma | 86.0 | 88.7 | 85.4 | 90.8 | 88.9 | 94.2 | 93.9 | 95.1 | 97.3 |
| ALL | 54.1 | 62.4 | 67.1 | 70.1 | 75.0 | 79.7 | 81.3 | 83.5 | 90.0 |
| AML | 18.7 | 26.2 | 26.4 | 31.7 | 36.9 | 41.0 | 39.4 | 48.4 | 65.1 |
| Neuroblastoma | 52.7 | 57.0 | 53.4 | 52.0 | 60.5 | 76.3 | 67.0 | 69.0 | 76.3 |
| NHL | 43.4 | 53.9 | 63.5 | 68.1 | 70.3 | 72.1 | 77.8 | 81.9 | 83.9 |
| Soft tissue | 65.2 | 68.8 | 68.2 | 72.4 | 67.2 | 68.9 | 73.8 | 70.9 | 78.1 |
| Wilms tumor | 72.6 | 78.0 | 86.5 | 91.0 | 92.2 | 91.3 | 91.5 | 92.1 | 91.5 |
Childhood acute lymphoblastic leukemia (ALL)
ALL is the most common malignancy in children in the United States. The peak incidence is between 2 and 6 years of age, and there is a slight male predominance. Stepwise gains in outcome in childhood ALL equal or surpass gains in other areas of childhood cancer have come through the results of large international cooperative groups. Refined risk-adapted approaches for therapy allocation and dose/schedule intensification of therapy have been primarily responsible for improving the overall survival rate to more than 90%.6
Although the exact etiology of childhood ALL is unclear, genetic factors play a significant role in the etiology of childhood ALL in selected cases. Various constitutional chromosomal abnormalities have been associated with pediatric ALL, such as DS, Bloom’s syndrome, Fanconi anemia, and ataxia telangiectasia. Of these, DS-ALL deserves special attention. Children with DS have 10–20 times higher risk of developing leukemia compared to children without DS. These children have unique clinical and biological characteristics that affect their treatment and outcome. Notably, T-cell and mature B-cell immunophenotype, and favorable cytogenetic features such as hyperdiploidy and the ETV6-RUNX1 translocation are not as common in this group of patients. Novel somatic alterations such as JAK2-activating mutations are found in about 20% of DS-ALL, while CRLF2 alterations (P2RY8-CRLF2 fusion) has been observed in about 50% of DS-ALL cases.7, 8 DS-ALL children are more prone to methotrexate-related toxicities, with more frequent mucositis and infectious complications. In general, prognosis of DS-ALL is worse than non-DS ALL, however, enhanced supportive measures have improved the outcome. More recently, genome-wide association studies (GWAS) have identified inherited genetic variation at the GATA3 locus being associated with ALL in adolescents and young adults (AYAs) population.9
Children with ALL usually presents with signs and symptoms suggestive of bone marrow infiltration, manifesting as fever (60%), bleeding symptoms (50%), and bone pain (30%). Child may present with a limp or refusal to walk. Lymphadenopathy and hepatosplenomegaly are present in about half to two-thirds of cases and is usually asymptomatic. Rarely, patients may present with symptoms suggestive of extramedullary spread, such as CNS involvement, testicular enlargement, and ocular changes. The diagnosis is typically made by bone marrow aspiration and/or biopsy. Multiple cell surface antigen markers, cytogenetic, and less commonly histochemical staining abnormalities in the leukemic cells are used to establish the diagnosis of ALL subtype and result in specific therapeutic decisions.10 About 85% of cases of childhood ALL express membrane markers consistent with B-precursor lineage, and about 15% express T-lineage markers. Although age and initial white blood cell (WBC) count at the time of disease presentation remain important predictors of outcome, other factors such as blast immunophenotype and cytogenetics, CNS involvement, and early response to chemotherapy are currently being utilized for risk stratification. Cytological confirmation of leukemic cells in cerebrospinal fluid (CSF) is required for the diagnosis of CNS leukemia. Standard or average risk patients are defined as age between 1 and 9 years with an initial WBC count of less than 50,000/mm3, while age more than 10 years with a presenting WBC of more than 50,000/mm3 are classified into high-risk disease category. Risk assessment is further refined by presence or absence of favorable [e.g., hyperdiploidy (specifically trisomies 4 and 10) or EVT6/RUNX1 fusion] and unfavorable cytogenetic features (e.g., hypodiploidy or iAMP21), overt CNS and testicular disease at diagnosis, and minimal residual disease (MRD) at day 8 and the end of induction chemotherapy. Philadelphia chromosome positive (ph+) ALL or t(9:22) occurs in 2–3% of pediatric ALL patients, and historically has been associated with a poor prognosis, now have much better outcome with the incorporation of tyrosine kinase inhibitors (TKI) in combination with chemotherapy.
Contemporary therapies include several components, namely, induction, presymptomatic, or prophylactic central nervous system therapy, postinduction intensification, and maintenance therapy. This design of current treatments will cure more than 80–90% of patients overall.6
Additional recent advances in therapy include the following:
- Current frontline therapy regimens are tailored to risk category as assessed by clinical features and biologic profile.10
- The substitution of dexamethasone for prednisone has improved the survival for standard risk patients, while no event-free survival (EFS) advantage has been noted for patients more than 10 years of age. In addition, there was significantly increased rate of osteonecrosis in patients more than 10 years of age.11
- Initial response to therapy determined by day 8 peripheral blood and/or bone marrow assessment of MRD at end induction (day 29) is an important predictor of outcome and is now utilized in risk-adjusted treatment approaches.12
- Postinduction intensification (or delayed intensification) improves outcome for both high and standard risk, B-precursor and T-lineage patients particularly when augmented regimens were utilized.13 However, there was no additional benefit of second delayed intensification.13
- Improved outcome associated with specific cytogenetic aberrations including hyperdiploidy, trisomy 4, 10, and t(12;21) (ETV6-RUNX1) and these patients are now classified as having low-risk disease, and receive less intensive chemotherapy if MRD is negative at day 8 and at day 29.14
- High-dose methotrexate given during interim maintenance improves outcome in high-risk patients.15
- Current systemic therapy allows elimination of cranial irradiation for almost all patients except those who have overt CNS disease at diagnosis.16
- Review of comparative outcome results of AYAs treated with pediatric or adult protocol-based therapy has shown improved outcomes for those treated utilizing the pediatric approach. However, treatment mortality comprises a larger percentage of adverse events than in younger children.17, 18
- Ph+ pediatric ALL can be treated with aggressive chemotherapy and concomitant targeted therapy with tyrosine kinase inhibitor—imatinib or dasatinib—leading to remarkable improvement in EFS without the need for stem cell transplantation.19
- Hematopoietic stem cell transplantation (HSCT) has a limited role in newly diagnosed ALL, except in refractory settings, those with hypodiploidy and possibly those patients with persistently high MRD at later time points. Early relapsed ALL (bone marrow relapse within 36 months from initial diagnosis or while in therapy) generally undergo HSCT, while late relapsed cases are treated with chemotherapy, and HSCT is considered on individual basis.
In addition to the disease- and treatment-related prognostic variables, recent studies have highlighted the importance of host factors in governing the disease outcome as well. Host factors refer to differences among patients with regard to drug absorption, metabolism, and sensitivity. Considerable heterogeneity has been described with respect to host pharmacology of thiopurines20 and vincristine.21 Furthermore, SNPs have been identified to be associated with unfavorable pharmacokinetics of various antileukemic chemotherapeutics, suggesting that drug disposition influences the risk of relapse.22
The heterogeneity of childhood ALL is reflected in emerging genomic and epigenomic studies. DNA copy number abnormalities were identified in genes that encode regulators of B-cell development, as CDKN2A/B, PAX5, IKZF1, and EBF1.23 In addition, somatic mutations in JAK2, alterations in RAS pathway genes (NRAS, KRAS, PTPN11, and NF1) and CRLF2 genomic alterations have been identified with increased frequency in high-risk patients.24, 25 JAK1 and JAK2 mutations have previously been shown to be mutated in DS-ALL and T-cell ALL as well.8 Furthermore, gene expression profiling has identified a subset of patients whose blasts share a gene expression profile observed in patients with Ph+ ALL but who lack the BCR-ABL1 fusion protein. Such patients are called “Ph-like” ALL.26 Ph-like ALL is associated with worse outcomes and deletions or mutations of IKZF1 are a characteristic feature of both Ph-positive and Ph-like ALL.27 This subgroup harbors tyrosine kinase fusions other than BCR-ABL, including ABL1, ABL2, CSF1R, and PDGFR fusions, and are candidates for treatment with tyrosine kinase inhibitors,28 while another subset of these patients harboring JAK mutations are candidates for JAK inhibitors.29 This approach is currently under evaluation in the high-risk ALL trial through COG.
Although significant advances have been made in the outcomes of newly diagnosed ALL over the past four decades, infant ALL and early relapsed ALL are the two subgroups whose outcome remains dismal, and there is an urgent need of innovative strategies for these patients. Because of increased FLT3 (a tyrosine kinase) expression in infant ALL, the incorporation of the FLT3 inhibitor lestaurtinib on the standard intensive chemotherapy backbone in infant leukemia is currently being investigated. Also given the unique biology of infant ALL with the majority of patients having rearrangement of the MLL gene on 11q23, the protein DOT1L is recruited to MLL target genes resulting in aberrant expression of genes. DOT1L inhibitors are in clinical trials.
For relapsed ALL, various novel treatment strategies are emerging including molecularly targeted agents and cellular therapies. Recently, bispecific T-cell engager antibodies have gained considerable attention. These proteins have both T-cell specific and B-cell specific regions, thereby directing the patient’s own cytotoxic T cells to the malignant tumor cells. Blinatumomab, a bispecific CD19-directed CD3 T-cell engager, has shown single agent activity and safety in children with multiple recurrent and refractory B-ALL, and is currently in clinical trial for children with B-ALL at first relapse through COG. Recent developments in cellular- and immune-based therapy have led to another major leap in the treatment of relapsed and refractory ALL. Chimeric antigen receptors (CARs)-modified T cells are genetically engineered to express antibodies directed against tumor antigens (CD19 for B-ALL) as well as T-cell activation molecules. They have shown great promise for long-term remission and possibly cure for this group of patients with extremely poor prognosis.30
Acute myeloid leukemia (AML) in children
AML comprises about 20% of pediatric leukemia and chronic myeloid leukemia is detected in about 1–2%. Similar to ALL, the development of AML has also been associated with various inherited and acquired predisposition syndromes, such as DS, bone marrow failure syndromes such as Fanconi anemia, dyskeratosis congenita, severe congenital neutropenia (Kostmann syndrome), Diamond–Blackfan anemia, as well as acquired aplastic anemia. Patients treated with chemotherapy (particularly alkylating agents and topoisomerase inhibitors) and radiation therapy are at risk of developing therapy-related AML (t-AML). The presentation of AML is similar to that of ALL; occasionally, child may present with violaceous, raised, plaque-like lesions on the skin or gums, called chloromas, which is an extramedullary manifestation of AML. Patients with acute promyelocytic leukemia (APL), a distinct AML subtype, characterized by t(15:17) or PML-RARA fusion, often present with severe coagulopathy and frequently have hyperleucocytosis.
Compared to ALL, pediatric AML is less responsive to available chemotherapy. AML survival has recently improved with cure rates approaching up to 60–70% in the pediatric population.31 This has been achieved by dose intensification, improved supportive measures, results from large clinical trials, and better understanding of the biology and heterogeneity of this disease. Unlike ALL, age and WBC are not considered as independent prognostic variables for stratification. In fact, cytogenetic and molecular characteristics and response to induction chemotherapy are utilized in current treatment schema for risk-adapted therapy.32, 33 Presence of inversion 16, t(8;21) and t(15:17), biallelic CEBPA mutations, and NPM1 mutations are considered as favorable, while monosomy 7, monosomy 5/del(5q), and FLT3-ITD with high allelic ratio are considered unfavorable characteristics. AML therapy consists of remission induction, prophylactic CNS-directed therapy, and postremission therapy. Chemotherapy regimens are intensive requiring prolonged hospitalizations and aggressive supportive measures. After initial 1–2 induction courses and upon achieving morphological remission (less than 5% blasts in marrow), postremission therapy is administered, which may comprise additional intensive chemotherapy or HSCT depending on the cytogenetic risk factors and the depth of response (MRD status) to induction chemotherapy. Prolonged maintenance courses have been shown to be inferior and are not incorporated in current regimens except in APL. APL treatment requires rapid initiation of all-trans retinoic acid (ATRA), a differentiation-inducing agent, with concomitant chemotherapy along with aggressive supportive measures, followed by prolonged maintenance phase consisting of ATRA, 6-mercaptopurine, and methotrexate. This regimen leads to the EFS of 70–80%, without the use of HSCT in first complete remission in APL.34 Arsenic trioxide has been shown as active agent against APL, and early experience in children with APL has shown encouraging results.
As the more intensive induction regimens have been maximally intensified, new active agents and strategies are needed. Currently, COG is studying in randomized study design the effect incorporating bortezomib, a proteasome inhibitor in FLT3-ITD-negative patients or sorafenib, a multitarget tyrosine kinase inhibitor that targets FLT3, c-KIT, PDGF, VEGF, and MEK/ERK/RAF pathway, for patients with high allelic ratio FLT3-ITD-positive AML, to the standard chemotherapy backbone for AML. In addition, epigenetic modifiers, such as vorinostat and 5-azacytadine, monoclonal antibodies against myeloid cell surface antigens, and various transplantation conditioning regimen and graft sources are currently being explored to improve the outcomes of pediatric AML.
Non-Hodgkin lymphoma in children (NHL)
Approximately 15% of all childhood cancers diagnosed in the United States are lymphomas. Sixty percent of all childhood lymphomas are classified as NHLs, representing 3% of all childhood malignancies for children younger than 5 years, and 8–9% for children and adolescents 5–19 years of age. In contrast to adults where most NHLs are low or intermediate grade, almost all NHL in children are high grade. There are three major subtypes of childhood NHL: (1) mature B-cell lymphoma predominantly Burkitt’s lymphoma (classic and atypical) or diffuse large B-cell lymphoma (DL-BCL), (2) precursor T-cell lymphoma or lymphoblastic lymphoma, and (3) mature T-cell or null cell lymphomas, anaplastic large-cell lymphoma (ALCL). The World Health Organization (WHO) classifies ALCL as a peripheral T-cell lymphoma. The distribution of these histologic subtypes includes approximately 40% Burkitt lymphoma, 30% lymphoblastic lymphoma, 20% diffuse large B cell, and 10% ALCL. Burkitt’s lymphoma tumor cells are characterized by a chromosomal translocation juxtaposing the c-myc oncogene and immunoglobulin locus regulatory elements t(8:14), rarely t(8:22) or t(2:8). Similarly, more than 90% of pediatric ALCL have a characteristic chromosomal rearrangement involving the ALK gene t(2:5).
Presence of bulky disease poses two potentially life-threatening situations that are often the presenting feature in children with NHL. First is the tumor lysis syndrome resulting in major electrolyte imbalances, most notably, hyperuricemia, hyperkalemia, and hyperphosphatemia. Aggressive hydration, allopurinol, or rasburicase (urate oxidase) are used in such situations along with other supportive measures. Second is the presence of large mediastinal masses especially seen in lymphoblastic lymphoma, posing a risk of cardiac or respiratory arrest (superior vena cava syndrome).
The primary modality of treatment of all histologic types and stages of childhood NHL is multiagent chemotherapy. The exact regimen of chemotherapy with or without intrathecal therapy and the intensity and length of treatment are usually dictated by the extent of disease and the histologic subtype. Although Burkitt’s lymphoma, DL-BCL, and ALCL are treated with short intensive courses of chemotherapy, lymphoblastic lymphoma is treated based on ALL regimens consisting of long maintenance phase. The role of surgery is critically important in the diagnosis and staging process, but it has a limited role in the overall treatment of childhood NHL. There is minimal to no role of radiation therapy in the overall treatment of childhood NHL.
The prognosis for children and adolescents with NHL, both with limited-stage and advanced-stage disease, has improved significantly over the past two decades. Except for rare subtypes, the chance of being alive and disease free at 5 years for limited-stage and advanced-stage disease B NHL is 95% and 80%, respectively.35 The prognosis for advanced lymphoblastic NHL in children and adolescents has now increased to over 85% survival.36 The prognosis, however, for the most advanced childhood and adolescent ALCL is still less than 70% at 7 years of follow-up.37
Recently, therapeutic strategies for childhood NHL are incorporating surface, intracellular, and molecular targets in order to not only improve the overall cure rates of advanced-stage disease but to minimize the collateral damage from the chemotherapy agents. Rituximab is a monoclonal antibody targeting the CD20 antigen, expressed by Burkitt’s lymphoma and DLBCL in children. The COG has demonstrated safety and tolerability of adding rituximab to the standard chemotherapy backbone in children and adolescents, with a 3-year EFS of 90% in patients with advanced mature B-cell lymphoma (with bone marrow and CNS involvement).38 Likewise, the promising activity of crizotinib (targeting ALK)39 and brentuximab vedotin (monoclonal anti-CD30 antibody)40 in relapsed ALCL has now led to a randomized phase 2 trial of adding these agents in combination with chemotherapy in newly diagnosed ALCL.
Hodgkin disease (HD)
HD accounts for approximately 5% of pediatric malignancies in developed countries. Reed–Strenberg (RS) cells (multinucleated giant cells) are the hallmark of HD, which are present in a background of inflammatory cells consisting of eosinophils, small lymphocytes, plasma cells, neutrophils, histiocytes, and fibroblasts. RS cells nearly always express CD30, while CD15 is expressed in approximately 70% of cases. Two broad pathologic categories of HD are classical Hodgkin’s lymphoma and nodular lymphocyte predominant. Classical Hodgkin’s lymphoma accounts for the majority of cases of childhood HD, while nodular lymphocyte predominant Hodgkin’s lymphoma occurs only in 5–10% cases.
The cure rate for pediatric patients with HD is greater than 90%. Standard therapy for pediatric patients with HD includes combination chemotherapy and low-dose involved-field radiotherapy (RT). Clinical research in pediatric HD aims to delineate minimal treatment necessary for cure and eliminate or minimize late sequelae of treatment.
PET scans have been demonstrated to be useful biomarkers of early response and useful in response-based therapy approaches to determine which patients would be eligible for treatment with chemotherapy alone or those that require intensification of therapy. The recently completed COG HD trial demonstrated successful omission of involved field RT without comprising outcome in intermediate-risk patients who show rapid response (PET-CT-negative disease), to two cycles of ABVE-PC (doxorubicin, bleomycin, vincristine, etoposide, prednisone, cyclophosphamide) and a complete response after four cycles.41 Management of relapsed/refractory HD remains challenging, and currently high-dose chemotherapy followed by autologous HSCT is considered standard of care for most patients. Recently, brentuximab vedotin (CD30-directed antibody drug conjugate) is being used in combination with chemotherapy for relapsed/refractory disease. Study of the biology of the RS cell in an effort to identify new therapeutic targets is also receiving significant attention.
Renal tumors
Wilms tumor
WT accounts for over 90% of renal tumors in children and 6% of childhood cancers in general. Today survival rates for all patients exceeds 90% and even patients presenting with metastatic disease have an event-free survival rate of 80% at 2 years.42 The typical clinical presentation is that of a well-appearing child who has an abdominal mass noted as an incidental finding. Over 90% of patients have a unilateral tumor with multicentric or bilateral tumors accounting for the remainder of cases. Patients with unilateral tumors present on average at 36.5 months (males) to 42.5 months (girls), while those with bilateral disease present earlier in life (33–35 months) reflecting a genetic predisposition.43
Many congenital syndromes are associated with WT and analyses of such cases led to the discovery of tumor suppressor genes involved in the pathogenesis of both inherited and sporadic cases. Children with the WAGR (WT, aniridia, genitourinary abnormalities, and mental retardation) syndrome have a greatly increased risk of WT, and this syndrome is associated with constitutional (germline) deletions at 11p13. Within the deleted region lies the WT1 gene responsible for the risk of WT (as well as PAX5 responsible for aniridia). Disease pathogenesis follows a classic two-hit model with the tumor having lost both WT1 alleles. Likewise, the Denys–Drash syndrome (pseudohermaphroditism, degenerative renal disease) is associated with constitutional mutations of WT1 and tumors contain mutations in both alleles. Somatic mutations of WT1 are seen in 10–20% of sporadic WT.44
A second tumor suppressor gene, WT2, has been mapped to 11p15.5. Abnormalities of this region are associated with another syndrome where the risk of WT is particularly high, the Beckwith–Wiedemann syndrome (macroglossia, omphalocele, visceromegaly, and hemihypertrophy). This region encodes a large number of imprinted genes including insulin-like growth factor 2 (IGF2) and H19, which encodes a noncoding RNA that acts as a tumor suppressor gene.45 Somatic loss of heterozygosity (LOH) of WT2 is the most common defect in sporadic WT occurring in up to 80% of cases. Other mutations are seen in WT including WTX (32%), CTNNB1 (encoding β-catenin, 15%), and T53 (5%).44
WT presents typically as a well-circumscribed heterogeneously enhancing mass originating from kidney (Figure 3a). Pathologically, WT typically shows a triphasic pattern composed of blastemal, epithelial, and stromal cell types, although in some cases only one or two of these cell types may be visualized. Anaplasia is characterized by markedly enlarged nuclei, pleomorphism, and polyploidy mitotic features, and these are associated with a poor prognosis. Anaplasia may be focal or diffuse and is seen in 5% of tumors.46 In the absence of anaplasia, WTs are classified as having “favorable histology.” Nephrogenic rests are precursor lesions and represent persistent nephroblastic tissue that has not undergone full differentiation.47 Patients with nephrogenic rests (30% sporadic and 100% bilateral WTs) may be at risk for the development of a subsequent WT. There are two types of nephrogenic rests based on location: perilobar and intralobar. Most nephrogenic rests undergo gradual resolution.
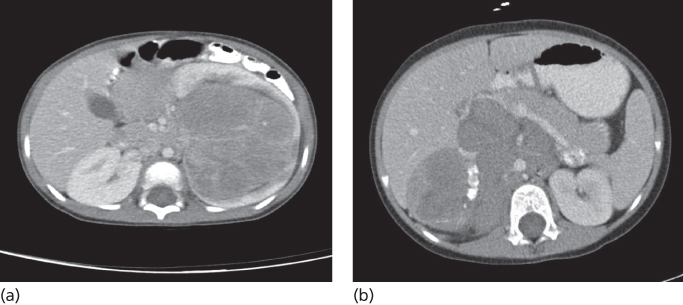
Figure 3 Computed tomographic (CT) imaging characteristics of common childhood abdominal tumors. (a) Wilm’s tumor: axial contrast-enhanced CT images of the abdomen showing a large well circumscribed heterogeneous left renal mass that does not cross the midline. (b) Neuroblastoma: axial contrast-enhanced CT images of the abdomen demonstrating a large lobulated heterogeneous mass in the right upper quadrant that crosses the midline with characteristic encasement of aorta and displacement of superior mesenteric vein, splenic vein, and pancreas anteriorly. Dense calcifications are present within the mass.
Therapy for WT involves surgical resection and chemotherapy with radiation reserved for more advanced stages. Worldwide there are two general approaches that lead to equivalent outcomes. In North America (based on practices established by the National Wilms Tumor Study Group (NWTSG) and now COG), most cases are treated by initial surgery (nephrectomy and lymph node sampling) followed by chemotherapy.42 In Europe (Societe Internationale d’Oncologie Pediatrique or SIOP), neoadjuvant therapy is advocated with delayed surgery. The advantage of the North American approach is that it allows for full, unaltered evaluation of all tumor tissue and accurate staging, while the SIOP approach allows for preoperative tumor shrinkage thereby making resection easier and decreasing the risk of spillage.48, 49
The usual definitive surgical approach to WT is a radical nephrectomy through a wide abdominal incision. The adrenal gland is removed and regional lymph nodes are sampled. More recently, a flank approach is being used and there is a question whether adrenalectomy is needed.50 Given the risk of second WTs, a partial nephrectomy or “nephron sparing” surgery is used for patients with bilateral tumors but also this is increasingly being considered for unilateral favorable histology tumors, especially in the neoadjuvant setting where there is assurance of negative margins due to an excellent response to chemotherapy. Finally, laparoscopic-/robotic-assisted resections are being explored by some surgeons.
Treatment of WT includes nephrectomy and stage-dependent chemotherapy with or without radiation to the abdomen and/or lungs. There are subtle differences in staging between the COG and SIOP approaches, but in general, complete resection of tumor with an intact capsule and no extension to the renal sinus is stage I. Stage II tumors are those completely resected, but the tumor extends beyond the capsule or into the renal sinus while incompletely resected tumors or those with lymph node involvement, tumor spillage, and/or positive surgical margins are declared stage III. Stage IV indicates metastatic hematogenous spread (usually lung but also liver, brain, or bone). Preoperative evaluation includes a CT of the abdomen and pelvis and Doppler ultrasound if there is concern about tumor thrombus in the inferior vena cava. Magnetic resonance imaging (MRI) may be useful for following nephrogenic rests and aid in surgical planning for patients with bilateral tumors.
Once removed, the tumors are evaluated pathologically to validate the diagnosis and examine sections for the presence of anaplasia. In addition, certain molecular markers may portend for a worse prognosis such as LOH at 1p and 16q, and these genetic lesions have been uniformly assessed on recent protocols.42
Patients less than 2 years with stage I favorable histology tumors weighing less than 550 g may be treated with nephrectomy only as the small minority who relapse can be salvaged with chemotherapy and RT. Patients with stage I and II tumors (without LOH at 1p and 16q) are treated with vincristine and dactinomycin (18 weeks), while stage III patients receive vincristine, dactinomycin, and doxorubicin with abdominal irradiation (24 weeks). Patients with LOH at 1p and 16q are upstaged and require additional therapy. Many different drug combinations have been assessed for favorable histology stage IV patients, but none have seemed to improve outcome over the three drug combination given with abdominal and chest irradiation. Those patients whose metastatic lesions regress early in therapy may not require chest XRT, whereas those patients whose lesions do not regress early require chest irradiation and may benefit from additional chemotherapy. The presence of anaplasia is associated with an inferior prognosis and therapy is modified based on whether the anaplasia is diffuse or focal as well as stage. For example, patients with stage II–IV tumors showing diffuse anaplasia are treated with combination therapy including vincristine, doxorubicin, cyclophosphamide, carboplatin, etoposide although the optimal regimen has yet to be established.
Patients with bilateral tumors and those with unilateral tumors and a congenital predisposition syndrome pose a special challenge as these patients are at risk for subsequent tumors. These patients can be treated with preoperative chemotherapy [vincristine, dactinomycin (unilateral tumors with predisposition syndrome) ±doxorubicin (bilateral tumors)] and undergo nephron sparing surgery at weeks 6 or 12 depending on optimal response. Such patients usually do not require an initial biopsy given the typical clinical and radiographic presentation. Therapy after resection is guided by stage and histology of the resected specimen.
The outcomes for all stages of favorable histology WT is excellent with 10-year relapse-free survival rates of 91% (stage I), 85% (stage II), 84% (stage III), and 75% (stage IV) while historically patients with anaplastic tumors have a worse outcome (10-year relapse-free survival 43% (stages I–III), 18% (stage IV)). Patients with bilateral tumors have an intermediate 10-year relapse-free outcome of approximately 65% (overall survival 78%).42
Other renal tumors
Renal cell carcinomas, clear cell sarcomas of the kidney and malignant rhabdoid tumors constitute 5.9%, 3.5%, and 1.6% of all renal tumors in children. Children with renal cell carcinoma present with abdominal pain, hematuria, and an abdominal mass on examination. Many conditions are associated with renal cell carcinoma including von Hippel–Lindau disease, familial renal cell carcinoma and previous therapy for malignancy. Pediatric renal cell carcinomas differ from adults in that a higher proportion are papillary and not otherwise specified, and the clear cell and chromophobe variants are less common.51, 52 Most tumors in childhood are characterized by translocations involving transcription factor E3 gene (TFE3) located on Xp11.2 with a variety of fusion partners. Children tend to present at a more advanced stage compared to adults. Radical nephrectomy with lymph node dissection remains the primary treatment. Survival rates at 4 years are 92%, 85%, and 73% for stages I, II, and II respectively while outcome for stage IV disease remains dismal (14%).42, 53 Optimal therapy for patients with unresectable or metastatic disease has not been defined but there is great interest in using tyrosine kinase inhibitors (sunitinib and others) in translocation positive renal cell carcinoma.42
Clear cell sarcomas are a distinct group of tumors with the histological appearance of nests of cells separated by organized fibrovascular septa. The molecular pathogenesis is unknown for most tumors although a t(10;17)(q22,p13) is noted in a small subgroup of these cases. Clear cell sarcomas can metastasize to bone and brain in addition to lung and liver but most patients present with localized disease. Historically these patients have been classified as unfavorable but incremental improvements have been achieved over successive NWTS trials. Patients treated on a regimen of vincristine, doxorubicin, cyclophosphamide, and etoposide with radiation therapy to the tumor bed had a 5 year EFS of 79%.42, 54
Malignant rhabdoid tumors present at a younger age compared to WT and symptoms include fever and gross hematuria.55 The majority of patients have advanced disease and up to 15% have associated CNS lesions. Histologically, the tumor is composed of large cells, prominent nucleoli, and eosinophilic cytoplasmic granules. These tumors are due to biallelic inactivation (mutation and/or deletion) of the SMARCB1 gene on 22q (also known as INI1) which encodes a member of a chromatin remodeling complex.56 About a third of patients harbor a germline mutation in SMARCB1 and these children present at an earlier age (median 5 months compared to 18 months). Most patients are treated with surgery, radiation, and multiagent chemotherapy but optimal drug combinations have yet to be identified. Young age, advanced stage and the presence of CNS disease are adverse prognostic factor. Four year relapse-free survival is 50%, 33%, 33%, and 21% for stages I, II, II, and IV respectively.42
Congenital mesoblastic nephroma typically occurs in infancy and present as unilateral abdominal tumors. Two patterns exist, classic and cellular. The cellular form of mesoblastic nephroma is characterized by the ETV6/NTRK3 fusion transcript (t(12;15)(p13;q25) identical to infantile fibrosarcoma.57 These tumors are treated by complete resection without need for adjuvant therapy since metastasis is quite rare. Overall survival rates are approximately 95%.58
Neuroblastoma
NB is the most common extracranial solid tumor of children and accounts for 8–10% of all childhood cancers. This tumor represents transformation of precursor neural crest cells that were destined to form sympathetic ganglia and the adrenal medulla. It is the most common tumor of infancy and the median age of presentation is 18 months.59 Curiously the disease has a wide clinical and biological spectrum ranging from spontaneous regression of widespread disease in infants to unrelenting, aggressive disease usually in older children. About 2% of cases are familial in nature and 50% of these patients carry germline mutations in the anaplastic lymphoma kinase gene, ALK, while a much smaller number of familial cases are due to mutations in Phox2b in association with other neurocristopathies like Hirschsprung’s disease.60–62 It is likely that additional steps are required for full transformation since only 50% of germline carriers of ALK mutations develop NB.63 Indeed GWAS have identified a number of predisposition loci yet their contribution to most sporadic cases is likely to be quite modest.64 ALK mutations and amplifications are seen in 8–12% and 2–3% respectively of sporadic cases and Phox2b mutations have been detected in 2% of sporadic tumors.65
The clinical presentation of NB is dependent on the location of the primary tumor but a majority of patients have symptoms due to widespread disease at initial diagnosis such as fever, weight loss, irritability, bone pain, and cytopenias. Two-thirds of cases are associated with an abdominal primary and the great majority of NBs are located in the adrenal gland (Figure 3b). Children may present with abdominal pain, constipation, and distention. A large abdominal mass that typically crosses the midline is detected on physical examination. Cervical and thoracic tumors (posterior mediastinum) can be associated with Horner syndrome. Presacral and paraspinal tumors may present with cord compression such as urinary retention, motor weakness, and clonus. Finally, NB may present with an unusual paraneoplastic syndrome characterized by opsoclonus myoclonus syndrome (OMS), where children typically manifest darting eye movements and myoclonic jerks with or without ataxia.66 Interestingly, OMS is often associated with favorable prognosis tumors, but unfortunately, neurological symptoms persist after effective treatment of the NB.
Initial evaluation of NB includes cross-sectional imaging with CT or MRI of the chest, abdomen and pelvis to determine local, regional (including extradural extension), and distant sites of disease. Metaiobenzlguanidine (MIBG) is a norepinephrine analogue selectively taken up by sympathetic nervous tissue and is used to assess disease location not picked up by CT or MRI. Most tumors are MIBG avid but for those that are not avid, FDG-PET may be used. Bilateral bone marrow aspirates and biopsies are required to detect bone marrow involvement. The diagnosis is confirmed by biopsy of the primary or distant site involvement, or characteristic tumor cell in the bone marrow with elevated levels of vanillylmandelic acid (VMA) and homovanillic acid (HVA) in the urine.
Histologically, tumors can display a range of cellular differentiation from ganglioneuroma (differentiated) to ganglioneuroblastoma to typical NB. Typically, the tumor is composed of small round blue cells with rosettes. Histologically, tumors can be classified as favorable or unfavorable based on age, the degree of differentiation (differentiated associated with better prognosis), Schwannian stromal content, and the karyorrhexis index [MKI, the number of mitosis and karyorrhexis (fragmented nuclei) per 5000 cells, less than 100 or 200 MKI (depending on stroma, age, and degree of differentiation) is associated with a better prognosis].67
An International Neuroblastoma Staging System (INSS) was developed to harmonize approaches and analyses of outcome worldwide (Table 3).68
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


