Fig. 24.1
Intact immunoglobulin and common antibody fragments. (a) Schematic representation of an intact immunoglobulin molecule. Each heavy chain (blue) consists of three constant domains (CH1-3) and the variable domain (VH). CH1 and CH2 are linked by the flexible hinge region, which forms two disulfide bonds with the hinge region of the complementary heavy chain. Each light chain (purple) consists of one constant domain (CL) and one variable domain (V L ) and is associated with the heavy chain through a disulfide bond proximal to the carboxy-termini of the two chains (COOH). The antigen-binding regions of the molecule (Ag Binding) are found at the amino-termini of the VH/VL pairs (NH2) and are circled in red. The Fc portion of the molecule, consisting of CH2–3, is indicated. Domain labels are constant throughout the figure. (b) The F(ab’)2 antibody fragment. Enzymatic digestion of intact immunoglobulin with pepsin results in the cleavage of the molecule at the hinge region, maintaining the disulfide bonds and yielding the F(ab’)2 fragment. (c) Papain cleaves the hinge region of intact immunoglobulin just above the disulfide bonds, generating two Fab fragments. Fab fragments can also be created through genetic manipulation. The heavy and light chains can associate non-covalently (right) or may maintain a disulfide bond near the carboxy-termini (left)
24.2.3 Reporter Labeling
There are a number of reporter molecules available for use in visualizing and even quantifying the binding of an Ab to its target [10]. One such class of reporters is the group of laser-activated fluorescent molecules called fluorophores or fluorochromes, commonly used in flow cytometry (see Sect. 24.8). Other reporters can be enzymatic and therefore depend on chemical reactions to be detected. For these reporters, the Abs are chemically linked, or conjugated, to an enzyme such as alkaline phosphatase (ALP) or horseradish peroxidase (HRP). An intense color is generated by the product created when these enzymes are incubated with chromogenic substrates, allowing measurement with a spectrophotometer. It is also possible to incubate these enzyme-linked Abs with a chemiluminescent substrate, the product of which gives off light, which can then be quantified by a number of different instruments and even captured on film.
A common third approach, often used to allow greater flexibility for the multiplexing of targets, includes biotin-conjugated Abs [11]. Biotin recognizes streptavidin with a high level of specificity and affinity, forming one of the strongest known non-covalent bonds. Streptavidin can be linked either to fluorophores or to enzymes like HRP and ALP, providing the flexibility to use a particular biotinylated Ab across multiple assay platforms. Similarly, within a single platform, the same biotinylated Ab can be used in multiple wells or tubes and, if necessary, be identified by different colors by using varied streptavidin-conjugated reporters, as with the multiple fluorophores used in flow cytometry [12].
24.2.4 Primary and Secondary Antibodies
Some diagnostic assay formats require the use of Ab pairs for detection (see Fig. 24.6b for a schematic representation). The first, or primary, Ab is specific for the target. A secondary reporter-conjugated Ab can be used in cases where the primary Ab does not include a reporter. Anti-species Abs, which are directed against immunoglobulin molecules produced by a different species, are commonly used as secondary Abs. For example, mouse immunoglobulin is injected into a goat to produce an immune response, resulting in a polyclonal goat anti-mouse Ab preparation that can be labeled with a reporter molecule. The goat anti-mouse Ab preparation is used to detect the presence of the primary mouse mAb wherever it may be bound to the target. However, in order to avoid possible cross-reactivity and to minimize the complexity of the assay, simpler assays in which the primary Ab is directly conjugated to a reporter are preferred when the assay system permits.
24.3 Immunoprecipitation
For many years, specific Abs have been used as a means to bind and concentrate targets in solution [13]. This process, known as immunoprecipitation (IP), involves the mixing and incubation of the specific Ab with a solution containing the molecule of interest (Fig. 24.2). After sufficient time to allow the Ab to bind the target, the Ab itself can be captured through binding to beads coated with bacterial protein A, protein G, or a mixture of both. The solution can then be centrifuged to pellet the beads at the bottom of the tube, allowing the supernatant to be transferred or discarded. Through this process, the target has been isolated and greatly concentrated and is now more readily detected.
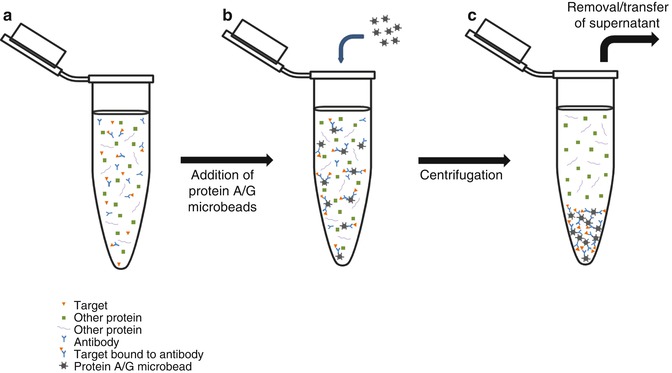
Fig. 24.2
Immunoprecipitation. (a) Cell lysate or other biological sample is incubated with specific antibody (Ab), which binds to the target in solution. (b) Microbeads coated with bacterial protein A, protein G, or a combination of both are added to the solution. The Abs, whether bound to target protein or free, will be bound by the bacterial proteins coating the bead. (c) Following centrifugation, the beads and their cargo of Ab and target protein will form a pellet at the bottom of the tube. The supernatant, now depleted of the target protein, can be transferred to another tube or discarded. These schematic representations of Abs and their targets will be used for all subsequent figures
When searching for comparatively rare proteins, which are present at much lower concentrations, a larger number of cells or volume of bodily fluids like plasma are required. This larger amount of material often presents problems for the detection system, which can be solved through the capture and concentration of the target by IP. In other cases, IP is used to diminish the amount of background detected by the assay system. The background can be minimized either by pulling the target out of the sample mixture for detection or by specifically depleting the mixture of an unwanted protein(s) that has been found to conflict with the detection of the target. IP is often used as a first step before detection by immunoblotting.
24.4 Immunoblotting
Also known as Western blotting, immunoblotting (IB) makes use of specific Abs for the detection of proteins of interest [14]. Sodium dodecyl sulfate (SDS) and heat are used to denature the proteins in a sample, which can range from a bodily fluid such as plasma, to a solution of cellular proteins released from cells by treatment with a lysis buffer. These proteins are separated according to mass via polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a membrane for detection (Fig. 24.3). The specific primary Ab is washed over the surface of the membrane for a prolonged incubation period, allowing it to bind the target protein, followed by incubation with a secondary enzyme-conjugated anti-species Ab. After the addition of a chemiluminescent substrate, a band of light will be generated at the position where the primary and secondary Abs are bound to the membrane. The amount of protein present dictates the amount of primary and secondary Ab bound to the membrane, which in turn dictates the intensity of the light generated. This light signal is traditionally detected by exposure to autoradiography film, but advances in low-light camera-based systems have led to increasing use of these documentation methods. On a traditional immunoblot exposed to film, lower-intensity signals correspond to fainter, thinner bands, while larger amounts of signal create fatter, darker bands (Fig. 24.3).
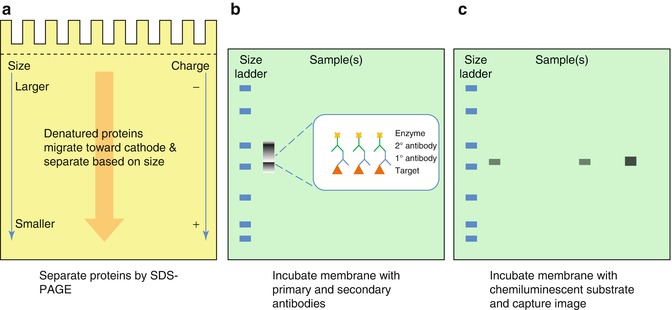
Fig. 24.3
Immunoblotting. (a) Samples are denatured in lysis buffer, loaded onto a polyacrylamide gel, and separated by electrophoresis (PAGE). The presence of sodium dodecyl sulfate (SDS) in the buffer masks the native charges of the proteins and lends an overall negative charge, allowing the proteins to migrate toward the cathode according to size, with smaller proteins traveling farther through the matrix than large proteins (SDS-PAGE). Proteins can also be analyzed by their native conformations under non-denaturing conditions in the absence of SDS (not shown). (b) Separated proteins are transferred to a nitrocellulose or polyvinylidene fluoride (PVDF) membrane via the application of electrical current. The membrane is then probed with primary Ab specific for the target protein or residue, followed by an enzyme-conjugated secondary anti-species Ab (more detail on secondary antibodies and reporters is given in Fig. 24.5). A molecular weight standard containing multiple proteins of known molecular weights is usually included in each experiment (size ladder), to provide an estimation of the distribution of the sample proteins. The proteins in these ladders are often dyed, sometimes with multiple colors, to allow visualization on the membrane. (c) The target is visualized by incubating the membrane with the chemiluminescent substrate of the reporter enzyme, which emits light. The signal is captured by exposure to autoradiography film or by a camera-based gel-documentation system. The quantity of target can then be extrapolated from signal intensity and/or band size, with larger bands corresponding to more bound target, although this measure is not truly quantitative, but relative to the other samples in that experiment only
Due to the fact that it provides an opportunity to physically view the interactions of an Ab with the proteins present in a sample matrix, immunoblotting is still widely used in a research setting despite being an older technique. This characteristic can help researchers determine the specificity of an Ab during the development of a cancer test, even if another technique will ultimately be used for detection. However, despite the fact that the method is comparatively time-consuming and labor intensive, there are still some cancer-related diagnostic tests which make use of Western blotting. Examples include confirmatory tests for Ri, Hu, or Yo, which are found in paraneoplastic syndromes associated with a number of cancers. The proteins of interest in these Western-based tests are actually Abs themselves. The Ri immunoblot detects the anti-Ri Ab present in patients with paraneoplastic myoclonus/opsoclonus syndrome, which is most often associated with gynecological cancers, breast cancer, and small cell lung cancer. The Yo, or Purkinje cell, Ab is also found in patients with breast, ovarian, and other gynecological cancers, in this case suffering from paraneoplastic cerebellar degeneration. Hu antineuronal nuclear Abs are detected by Western blot in a small percentage of patients with small cell lung cancer and are associated with paraneoplastic sensory neuropathy and encephalomyelitis. The highly specific Abs used in these Western blots provide confirmation of the identity of the Hu, Yo, and Ri Abs initially detected by first-line screening tests.
24.5 Radioimmunoassays
One of the first highly sensitive methods for measuring the levels of proteins such as hormones in the blood was the radioimmunoassay (RIA) [15]. In a classic RIA, a known quantity of purified target protein is radiolabeled, most often with a gamma radioisotope of iodine. This “hot” protein is mixed with a specific Ab that has been immobilized on a surface, and then the biological sample containing unlabeled or “cold” protein is added to the mixture (Fig. 24.4). In a standard competition assay, the cold protein will then compete with the radiolabeled protein for binding to the Ab, leading to the displacement of a fraction of the radiolabeled protein. The amount of target protein present in the sample can then be extrapolated by measuring the amount of displaced radioactivity.
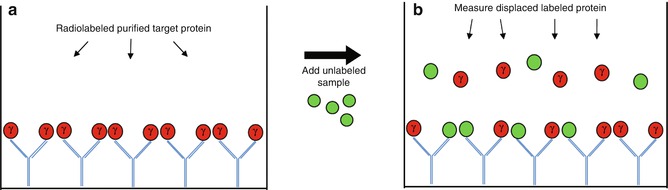
Fig. 24.4
Radioimmunoassay. (a) Purified target protein is radiolabeled, often with the gamma isotope of iodine (γ) and incubated with immobilized specific antibody (Ab). Sample containing unlabeled target protein is then added to the well. (b) The unlabeled target protein competes with the purified radiolabeled protein for binding to the Abs, displacing some of the radiolabeled protein when present at high enough concentrations. The unbound protein is removed from the well, and the radioactivity of the displaced radiolabeled protein is measured to give an indirect measure of the amount of unlabeled target protein present in the sample
RIA technology allowed some of the first specific and sensitive tracking of important hormones like insulin in human blood [16] and is still used in some cancer-related diagnostics today, including thyroid hormone testing. Some thyroid hormone tests, including reverse T3, free T4, and especially thyroid-stimulating hormone (TSH), are still offered via RIA. These thyroid hormone tests are included as diagnostic tests in the preliminary characterization of thyroid nodules as malignant or benign and in the diagnosis of TSH-secreting pituitary adenomas. In the interest of laboratory safety, however, technology has moved away from techniques requiring the handling of radioactivity, and the RIA method has largely been replaced by enzymatic immunoassays.
24.6 Enzymatic Immunoassays
Enzymatic immunoassays, or EIAs, are the archetypal antibody-based detection format and a foundation of basic cellular biology research. The best known EIA format is the enzyme-linked immunosorbent assay (ELISA) [17], which has been used for the detection of targets in both cell lysates and in nearly every bodily fluid, ranging from whole blood to sputum to cerebrospinal fluid. Most commonly, ELISA assays are performed in microtiter plates containing 96 or more wells, providing the opportunity to test a large number of samples in a single run. Further, as the treatment of each well is often identical, the format of the ELISA assay lends itself to a high degree of automation using liquid handling robots and plate washers. Since the ELISA often contains multiple lengthy incubation steps, the ease with which it can be automated provides valuable time and labor savings in a high-throughput cancer diagnostics laboratory.
ELISA formats can range from simple to complex, incorporating from one to four Abs (Fig. 24.5) [17]. At the most basic end of the spectrum is the “direct” ELISA, which uses a single reporter-labeled primary Ab to detect the target that has been adsorbed to the surface of the well or plate (Fig. 24.5a). More commonly used, however, is the “sandwich” ELISA, which can use from two to four Abs as shown in Fig. 24.5b. In many cases the sandwich format is preferred due to the greater level of specificity conferred by requiring two different specific antibodies to bind the target before detection is achieved. The first Ab which binds the target is referred to as the “capture” Ab and is bound to the plate/well either through direct adsorption or through interaction with a corresponding anti-species Ab that is bound to the plate instead. The capture Ab will bind the target during incubation with the lysate or bodily fluid, after which the irrelevant proteins are washed away, leaving the enriched and purified target. The second, or “detection,” Ab is now incubated in the well and allowed to bind to the target wherever it has been captured in the well. The detection Ab can be directly labeled with a reporter or can be detected itself by a secondary reporter-conjugated anti-species Ab. The important consideration to remember when designing a sandwich ELISA is that if a secondary anti-species Ab will be used for detection, the capture and detection Abs must have been generated in different species, to prevent the binding of the secondary detection Ab to both.
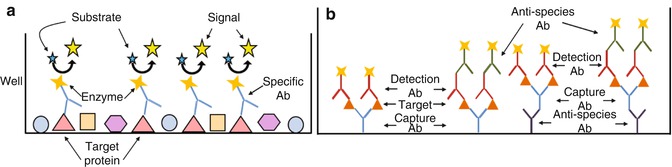
Fig. 24.5
ELISA. (a) The simplest ELISA consists of proteins adsorbed to the surface of a well and incubated with specific enzyme-conjugated Abs. After binding of the Abs to the target protein, the well is washed, and the colorimetric or chemiluminescent substrate is added. The reporter enzyme acts on the substrate, generating signal in the form of color or light, respectively. (b) The sandwich ELISA and its possible variations. The specific capture Ab can be directly coated onto the surface of the well or be bound itself by an anti-species Ab. After capture of the target protein, the target is bound by the detection Ab, which can be conjugated to a reporter itself or bound by a reporter-conjugated secondary anti-species Ab. Each of these permutations is represented
The flexibility made possible by the sandwich ELISA allows the detection of specialized protein motifs. Examples include the differentiation between isoforms created by alternative splicing [18] or detection of posttranslational modifications such as phosphorylation, acetylation, glycosylation, methylation, ubiquitination, and even protein cleavage [18–23]. The turnover rate of important proteins, the activation status of specific pathways, and other important cellular activities can be inferred from the posttranslational modifications of important cell signaling proteins. For detection of these modifications, the target protein can be bound by the capture Ab, the unbound background protein is washed away, and then a detection antibody specific for the modification of interest can be used to determine whether the protein contains that posttranslational change. The opposite approach can also be taken, in which a detection Ab specific for the target protein can be used to probe the proteins pulled out of solution by a capture Ab specific for phosphotyrosine, for example. In some cases, the posttranslational modification at a specific amino acid residue is even included in the immunogen, in order to generate an Ab specific ONLY for the version of the protein containing a phosphorylated residue at a given position rather than the non-phosphorylated version.
It is also theoretically possible, though generally technically difficult, to use a sandwich ELISA to detect the protein product of a gene fusion, such often happens in cancer. One such example is the BCR-ABL fusion protein which is the result of the so-called Philadelphia chromosome, or the reciprocal translocation t(9;22);(q34;q11), that occurs most often in chronic myeloid leukemia (CML). In this example, a capture Ab specific for the BCR protein would immobilize both wild-type (WT) and fused BCR, while only the fusion protein would be bound by the anti-Abl detection Ab.
The ability to detect multiple targets side by side in a single aliquot of sample can provide a great deal of important information, as well as maximize the information derived from the often inadequate and precious samples received in cancer diagnostic laboratories. Newer ELISA technologies have emerged in the last decade that make multiplexing possible through the use of multi-spot wells. In this assay layout, a number of different capture Abs are bound to the bottom of each well in discrete spots, ranging from 2 to 4 up to 100 (Fig. 24.6a). Flexibility has been further increased by breakthroughs in chemical linkers, which allow assay designers to mix and match the capture Abs in a given well and do it in-house (Fig. 24.6b). These linker-conjugated capture Abs are used with specialized plates, in which the binding partner of each chemical linker has already been spotted in a specific position on the bottom of the well. Each capture Ab will therefore only bind to one particular spot within the well, and the sample can then be added to the well and interrogated for the presence of many target proteins at once.
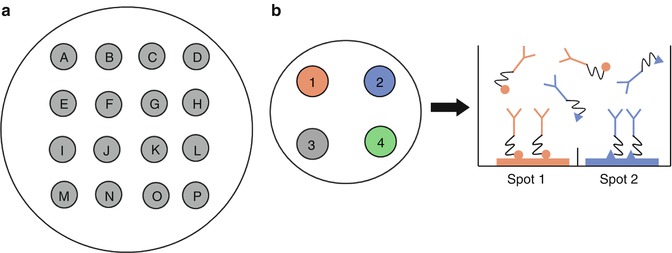
Fig. 24.6
Multi-spot ELISAs. (a) A schematic representation of a 16-spot multi-spot ELISA well. Each spot, or letter, corresponds to a different capture Ab that is carefully applied to the plate in one discrete area, usually by robot. A single sample can then be incubated in the well and 16 different sandwich ELISAs performed simultaneously on one small volume of analyte. (b) Chemical linkers can create multi-spot assays without robotic spotting of the capture antibodies, allowing mixing and matching of desired analytes. Each capture Ab is conjugated to one of several chemical linkers and incubated simultaneously in the well. Each linker binds only to its corresponding spot, isolating each capture Ab in one specific region of the plate. Multiple sandwich ELISAs can then be performed as in (a)
These sorts of multiplexed ELISA platforms generally require camera-based detection systems that include sophisticated software capable of discriminating and parsing the signal generated by multiple spots in a single small well. Adding an ever greater level of control over the process, some more advanced ELISA platforms now include computer-controlled initiation of the chemiluminescent reaction. In this system the reporter is a true electrochemiluminescent (ECL) reagent, requiring an electrical current to undergo the chemical reaction, and the assay is performed in a specialized plate containing a small electrode in each well. The computer controls the application of current, usually breaking the plate down into sections read in sequence. These sorts of adaptations to the ELISA platform represent some of the advances made in the last decade and will likely see increasing uptake in the design of cancer tests.
This versatility in the sandwich ELISA platform, as well as the flexibility provided by the large number of available reporter/detection formats, suggests that similarly ingenious ELISAs will continue to be developed. Most commonly in cancer diagnostics, however, more straightforward sandwich ELISAs are used for the purposes of quantitative detection and monitoring of relevant proteins. An example is the HER2 ELISA, which measures the level of HER2/neu present in the serum of breast cancer patients. With the inclusion of a standard curve on the ELISA plate, the amount of HER2/neu protein present in the well can be quantified, and the concentration of the protein circulating in the body can be extrapolated. These data can be used by the clinician to assess the patient’s prognosis and to determine the likely response of the patient to a given therapy. Further, if a baseline concentration of the circulating protein is established prior to administering therapy, subsequent longitudinal measurements can be compared to that baseline and used to monitor the efficacy of therapy.
Related posts:
 Role of Innate Immunity in Cancers and Antitumor Response
Role of Innate Immunity in Cancers and Antitumor Response
 Prognostic Value of Innate and Adaptive Immunity in Cancers
Prognostic Value of Innate and Adaptive Immunity in Cancers
 Nutrition, Immunity, and Cancers
Nutrition, Immunity, and Cancers
 Flow Cytometry in Cancer Immunotherapy: Applications, Quality Assurance, and Future
Flow Cytometry in Cancer Immunotherapy: Applications, Quality Assurance, and Future
 B Cells in Cancer Immunology: For or Against Cancer Growth?
B Cells in Cancer Immunology: For or Against Cancer Growth?
 Immunohistochemistry of Cancers
Immunohistochemistry of Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


