Fig. 4.1
(a) Schematic representation of the structure of the human glucocorticoid receptor (hGR) gene. Alternative splicing of the primary transcript gives rise to the two mRNA and protein isoforms, hGRα and hGRβ. (b) Functional domains of the hGRα. The functional domains and Fig. 4.1 (continued) subdomains are indicated beneath the linearized protein structures. AF activation function; DBD DNA-binding domain; LBD ligand-binding domain; NLS nuclear localization signal. (c) Enlargement of part of the DBD showing the amino acid sequence (single letter codes) of the two zinc fingers and the dimerization loop (in bold). The A to T mutation at position 458 that could produce a dimerization defective receptor is shown
The human GR is a modular protein composed of distinct regions illustrated in Fig. 4.1b: the amino-terminal A/B region, also called immunogenic or N-terminal domain (NTD), and the C, D, and E regions, which correspond to the DNA-binding domain (DBD), the hinge region, and the ligand-binding domain (LBD), respectively.
The NTD of the hGRα contains a major transactivation domain, termed activation function (AF)-1, which is located between amino acids 77 and 262 of the hGRα and is ligand-independent. The AF-1 plays an important role in the interaction of the receptor with molecules necessary for the initiation of transcription, such as coactivators, chromatin modulators, and basal transcription factors, including RNA polymerase II, TATA-binding protein (TBP), and a host of TBP-associated proteins (TAFIIs) [3, 5, 6].
The DBD of the hGRα corresponds to amino acids 420–480 and contains two zinc finger motifs through which the hGRα binds to specific DNA sequences, the glucocorticoid response elements (GREs) in the promoter region(s) of target genes [5, 6]. The DBD is the most highly conserved domain throughout the steroid receptor family. The two zinc finger motifs are able to tetrahedrally coordinate a zinc atom and are held by four cysteine (Cys) residues (Fig. 4.1c). Only very few amino acids, termed the proximal (P)-box, within the first zinc finger are responsible for specific recognition of the cognate GREs. Another set of amino acids, called the distal (D)-box within the second zinc finger, forms the weak dimerization interface of the DBD. The DBD of the hGRα also contains sequences important for nuclear translocation [5, 6].
The hinge region or region D is a flexible region located between the DBD and LBD. Its amino terminus is an integral part of the DBD and is involved in its dimerization. The hinge region confers structural flexibility in the receptor dimmers, thereby allowing a single receptor dimmer to interact with multiple GREs [5, 6].
The LBD of the hGRα corresponds to amino acids 481–777, binds to glucocorticoids, and plays a critical role in the ligand-induced activation of hGRα. The LBD also contains a second transactivation domain, termed AF-2, which is ligand-dependent, as well as sequences important for receptor dimerization, nuclear translocation, binding to the heat shock proteins (HSPs), and interaction with coactivators [5, 6].
Expressed hGRα is a panel of eight amino-terminal translational isoforms of varying lengths, each of which consists of three subdomains, the NTD, the DBD, and the LBD. These hGRα isoforms differ at their amino-termini and may differentially transduce the glucocorticoid signal to target tissues depending on their selective relative expression and inherent activities. It is likely that similar differential cell-specific production and functional differences might also be present between the putative hGRβ translational isoforms [5, 6]. This marked complexity in the transcription/translation of the hGR gene enables target tissues to differentially respond to circulating glucocorticoid concentrations and accounts for the highly stochastic nature of the glucocorticoid signaling pathway [11].
In the absence of ligand, hGRα resides mostly in the cytoplasm of cells as part of a hetero-oligomeric complex, which contains chaperon HSPs 90, 70 and FKBP51, as well as other proteins [7, 11]. Upon ligand-induced activation, the hGRα dissociates from this multiprotein complex and translocates into the nucleus, where it binds as a homodimer to GREs in the promoter regions of target genes and regulates their expression positively or negatively, depending on GRE sequence and promoter context [7, 11] (Fig. 4.2). To initiate transcription, the hGRα uses its transcriptional activation domains, activation function (AF)-1 and AF-2, located in the NTD and LBD, respectively, as surfaces to interact with nuclear receptor coactivators and chromatin-remodeling complexes [12–15]. The ligand-activated hGRα can also modulate gene expression independently of DNA-binding, by interacting, possibly as a monomer, with other transcription factors, such as nuclear factor-κB, activator protein-1, p53, and signal transducers and activators of transcription [7]. Following transcriptional activation or inhibition of glucocorticoid-responsive genes, the hGRα dissociates from the ligand and has a lower affinity for binding to GREs. The unliganded hGRα remains within the nucleus for a considerable length of time and is then exported to the cytoplasm; both within the nucleus and within the cytoplasm the hGRα may be recycled and/or degraded in the proteasome [16] (Fig. 4.2).
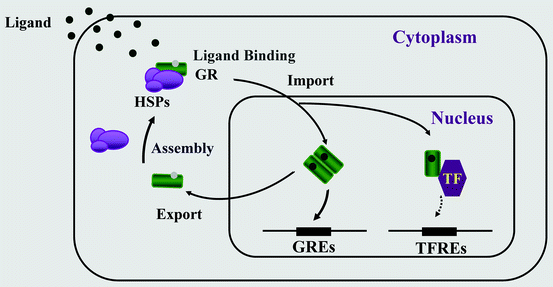
Fig. 4.2
Nucleocytoplasmic shuttling of the glucocorticoid receptor. Upon binding to the ligand, the activated hGRα dissociates from heat shock proteins (HSPs) and translocates into the nucleus, where it homodimerizes and binds to glucocorticoid response elements (GREs) in the promoter region of target genes or interacts with other transcription factors (TFs), such as activator protein-1 (AP-1), nuclear factor-κB (NF-κB), and signal transducer and activator of transcription-5 (STAT5), ultimately modulating the transcriptional activity of respectively GRE- or TFRE-containing genes
Alterations in the molecular mechanisms of hGRα action may lead to alterations in tissue sensitivity to glucocorticoids, which may take the form of glucocorticoid resistance or glucocorticoid hypersensitivity and may be associated with significant morbidity [17–19] (Table 4.1). In the present review, we summarize the pathophysiology and molecular mechanisms underlying Chrousos syndrome and primary generalized glucocorticoid hypersensitivity.
Table 4.1
Expected clinical manifestations in tissue-specific glucocorticoid resistance or hypersensitivitya
Target tissue | Glucocorticoid hypersensitivity = glucocorticoid excess | Glucocorticoid resistance = glucocorticoid deficiency |
|---|---|---|
Central nervous system | Insomnia, anxiety, depression, defective cognition | Fatigue, somnolence, malaise, defective cognition |
Liver | + Gluconeogenesis, + lipogenesis | Hypoglycemia, resistance to diabetes mellitus |
Fat | Accumulation of visceral fat (metabolic syndrome) | Loss of weight, resistance to weight gain |
Blood vessels | Hypertension | Hypotension |
Bone | Stunted growth, osteoporosis | |
Inflammation/immunity | Immune suppression, anti-inflammation, vulnerability to certain infections and tumors | + Inflammation, + autoimmunity, + allergy |
Primary Generalized Glucocorticoid Resistance and Hypersensitivity
Clinical Manifestations
Primary generalized familial and sporadic glucocorticoid resistance is a rare, familial or sporadic condition, initially described and elucidated by Chrousos et al. This condition is characterized by generalized, mostly partial, target-tissue insensitivity to glucocorticoids, which leads to compensatory activation of the hypothalamic-pituitary-adrenal (HPA) axis and hypersecretion of adrenocorticotropic hormone (ACTH) in the systemic circulation [20–22]. The latter results in adrenocortical hyperplasia, increased cortisol secretion as a compensation for the reduced action of glucocorticoids at target tissues, and increased production of adrenal steroids with mineralocorticoid [cortisol, deoxycorticosterone (DOC) and corticosterone] and/or androgenic activity [androstenedione, dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEAS)] [20–22].
The clinical manifestations of PGGR reflect the pathophysiologic alterations described above and primarily include those of mineralocorticoid and/or androgen excess [20–22]. Clinical manifestations of glucocorticoid deficiency might occur, but are rare and were only reported in a young child with hypoglycemic generalized tonic-clonic seizures during the course of a febrile illness [23], in a newborn baby with severe hypoglycemia, excessive fatigability with feeding, increased susceptibility to infections and concurrent growth hormone deficiency [24], and in several adult patients with chronic fatigue [20–22]. Clinical manifestations of mineralocorticoid excess include hypertension and hypokalemic alkalosis. Clinical manifestations of androgen excess include ambiguous genitalia in a karyotypic female at birth and gonadotropin-independent precocious puberty in children of either gender; acne, hirsutism, and hypofertility in both sexes; male-pattern hair loss, menstrual irregularities, and oligo-anovulation in females; and oligospermia in males [20–22]. The clinical spectrum of the condition is broad, ranging from most severe to mild forms, while a number of patients may be asymptomatic, displaying biochemical alterations only [20–22]. This variable clinical phenotype is due to variations in the tissue sensitivity of the glucocorticoid, mineralocorticoid, and/or androgen receptor signaling pathways; variations in the activity of key hormone-inactivating or -activating enzymes, such as the 11β-hydroxysteroid dehydrogenase [25] and other genetic or epigenetic factors, such as the presence of insulin resistance and visceral obesity [21]. In recognition of Professor George P. Chrousos’ extensive and ground-breaking research work in this field, it has been proposed that the term “Chrousos Syndrome” is used in place of “Primary Generalized Familial and Sporadic Glucocorticoid Resistance” [26, 27].
Primary generalized glucocorticoid hypersensitivity (PGGH) represents the mirror image of Chrousos syndrome and is characterized by generalized, partial, target-tissue hypersensitivity to glucocorticoids, and compensatory hypoactivation of the HPA axis. To date, there has been only one patient reported with manifestations of tissue-specific glucocorticoid hypersensitivity caused by a novel hGR gene mutation. The patient was a 43-year-old female, who presented with a long-standing history of visceral obesity, hypercholesterolemia, hypertriglyceridemia, diabetes type 2, and hypertension [28] (Table 4.2).
Table 4.2
Mutations of the human glucocorticoid receptor gene causing primary generalized glucocorticoid resistance or hypersensitivitya
Mutation position | |||||
|---|---|---|---|---|---|
References | cDNA | Amino acid | Molecular mechanisms | Genotype | Phenotype |
Chrousos et al. [20] | 1922 (A → T) | 641 (D → V) | Transactivation ↓ | Homozygous | Hypertension |
Hurley et al. [30] | Affinity for ligand ↓ (×3) | Hypokalemic alkalosis | |||
Nuclear translocation: 22 min | |||||
Abnormal interaction with GRIP1 | |||||
Karl et al. [31] | 4 bp deletion in exon-intron 6 | hGRα number: 50% of control | Heterozygous | Hirsutism | |
Inactivation of the affected allele | Male-pattern hair-loss | ||||
Menstrual irregularities | |||||
Malchoff et al. [32] | 2185 (G → A) | 729 (V → I) | Transactivation ↓ | Homozygous | Precocious puberty |
Affinity for ligand ↓ (×2) | Hyperandrogenism | ||||
Nuclear translocation: 120 min | |||||
Abnormal interaction with GRIP1 | |||||
Karl et al. [29] | 1676 (T → A) | 559 (I → N) | Transactivation ↓ | Heterozygous | Hypertension |
Kino et al. [33] | Decrease in hGR binding sites | Oligospermia | |||
Transdominance (+) | Infertility | ||||
Nuclear translocation: 180 | |||||
Abnormal interaction with GRIP1 | |||||
Ruiz et al. [34] | 1430 (G → A) | 477 (R → H) | Transactivation ↓ | Heterozygous | Hirsutism |
Charmandari et al. [39] | No DNA-binding | Fatigue | |||
Nuclear translocation: 20 min | Hypertension | ||||
Ruiz et al. [34] | 2035 (G → A) | 679 (G → S) | Transactivation ↓ | Heterozygous | Hirsutism |
Charmandari et al. [39] | Affinity for ligand ↓ (×2) | Fatigue | |||
Nuclear translocation: 30 min | Hypertension | ||||
Abnormal interaction with GRIP1 | |||||
Mendonca et al. [35] | 1712 (T → C) | 571 (V → A) | Transactivation ↓ | Homozygous | Ambiguous genitalia |
Affinity for ligand ↓ (×6) | Hypertension | ||||
Nuclear translocation: 25 min | Hypokalemia | ||||
Abnormal interaction with GRIP1 | Hyperandrogenism | ||||
Vottero et al. [36] | 2241 (T → G) | 747 (I → M) | Transactivation ↓ | Heterozygous | Cystic acne |
Transdominance (+) | Hirsutism | ||||
Affinity for ligand ↓ (×2) | Oligo-amenorrhea | ||||
Nuclear translocation ↓ | |||||
Abnormal interaction with GRIP1 | |||||
Charmandari et al. [38] | 2318 (T → C) | 773 (L → P) | Transactivation ↓ | Heterozygous | Fatigue |
Transdominance (+) | Anxiety | ||||
Affinity for ligand ↓ (×2.6) | Acne | ||||
Nuclear translocation: 30 min | Hirsutism | ||||
Abnormal interaction with GRIP1 | Hypertension | ||||
Charmandari et al. [40] | 2209 (T → C) | 737 (F → L) | Transactivation ↓ | Heterozygous | Hypertension |
Transdominance (time-dependent) (+) | Hypokalemia | ||||
Affinity for ligand ↓ (×\1.5) | |||||
Nuclear translocation: 180 min | |||||
Nader et al. [23] | 2141 (G → A) | 714 (R → Q) | Transactivation ↓ | Heterozygous | Hypoglycemia |
Transdominance (+) | Hypokalemia | ||||
Affinity for ligand ↓ (×2) | Hypertension | ||||
Nuclear translocation ↓ | Mild clitoromegaly | ||||
Abnormal interaction with GRIP1 | Advanced bone age | ||||
Precocious pubarche | |||||
McMahon et al. [24] | 2 bp deletion at nt 2318-9 | 773 | Transactivation ↓ | Homozygous | Hypoglycemia |
Affinity for ligand: absent | Fatigability with feeding | ||||
No suppression of IL-6 | Hypertension | ||||
Zhu Hui-juan et al. [41] | 1667 (G → T) | 556 (T → I) | Not studied yet | Heterozygous | Adrenal incidentaloma |
Charmandari et al. [28] | 1201 (G → C) | 401 (D → H) | Transactivation ↑ | Heterozygous | Visceral obesity |
Transdominance (+) | Hypercholesterolemia | ||||
Affinity for ligand: N | Hypertriglyceridemia | ||||
Nuclear translocation: N | Hypertension | ||||
Interaction with GRIP1: N | Diabetes type 2 | ||||
Molecular Mechanisms
hGR Mutations
The molecular basis of Chrousos syndrome has been ascribed primarily to mutations in the hGR gene, which impair the molecular mechanisms of hGR action and decrease tissue sensitivity to glucocorticoids (Table 4.2, Fig. 4.3) [23, 24, 29–41]. The molecular defects that have been elucidated in cases with Chrousos syndrome and have been reported to date are summarized in Table 4.2. We have identified most hGR mutations associated with primary generalized glucocorticoid resistance and have systematically investigated the molecular mechanisms through which these various natural hGR mutants affect glucocorticoid signal transduction in almost all reported cases of generalized glucocorticoid resistance. We studied: (a) the transcriptional activity of the mutant receptors; (b) the ability of the mutant receptors to exert a dominant negative effect upon the wild-type receptor; (c) the affinity of the mutant receptors for the ligand; (d) the subcellular localization of the mutant receptors and their nuclear translocation following exposure to the ligand; (e) the ability of the mutant receptors to bind to GREs; and (f) the interaction of the mutant receptors with the glucocorticoid receptor-interacting protein-1 (GRIP1) coactivator, which belongs to the p160 family of nuclear receptor coactivators and plays an important role in the hGRα-mediated transactivation of glucocorticoid-responsive genes [22, 29–40].
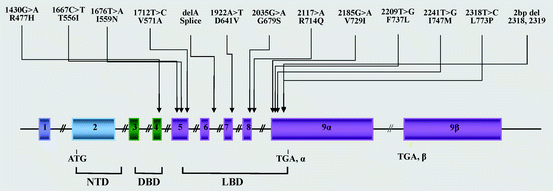
Fig. 4.3
Location of the known mutations of the hGR gene causing Chrousos syndrome.DBD: DNA-binding domain; LBD: ligand-binding domain; NTD: amino terminal domain
Compared with the wild-type receptor, all mutant receptors demonstrated variable reduction in their ability to transactivate glucocorticoid-responsive genes in response to dexamethasone [29–40]. The mutant receptors hGRαI559N, hGRαF737L, hGRαI747M, and hGRαL773P exerted a dominant negative effect upon the wild-type receptor, which might have contributed to manifestation of the disease at the heterozygote state [29, 33, 35, 37, 40]. All mutant receptors in which the mutations were located in the LBD of the receptor showed a variable reduction in their affinity for the ligand [29–40]. The only mutant receptor that demonstrated normal affinity for the ligand was the hGRαR477H, in which the mutation was located in the DBD [39]. Most pathologic mutant receptors were observed primarily in the cytoplasm of cells in the absence of ligand, except for the hGRαV729I and hGRαF737L receptors, which were localized both in the cytoplasm and the nucleus of cells. Exposure to dexamethasone induced a slow translocation of the mutant receptors into the nucleus, which ranged from 20 to 180 min compared with the wild-type hGRα, which required only 12 min for complete translocation [29–40]. All mutant receptors in which the mutations were located in the LBD preserved their ability to bind to DNA and displayed an abnormal interaction with the GRIP1 coactivator in vitro [29–40]. The only mutant receptor that failed to bind to DNA but displayed a normal interaction with the GRIP1 coactivator was the hGRαR477H, in which the mutation was located at the C-terminal zinc finger of the DBD [39].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


