Primary Brain Tumors
INTRODUCTION
Over 100 types of primary brain tumor are defined by the World Health Organization (WHO), the most commonly accepted classification system (1). Only a few entities account for the bulk of the incidence while the others are rare. The incidence rate of all primary central nervous system (CNS) tumors in the United States (U.S.) is 19.3 cases per 100,000 person-years (2). An estimated 64,530 primary CNS tumors were diagnosed in the United States in 2011. Approximately 24,070 of these tumors were malignant and this subgroup represented 1.46% of all malignant cancers diagnosed in the United States (2, 3).
Although relatively uncommon, primary brain tumors cause a disproportionate amount of morbidity and mortality, with an estimated 14,080 deaths attributed to malignant primary CNS tumors in the United States in 2013 (2). In this chapter we discuss the clinical evaluation of a patient with a suspected primary brain tumor and review the most common adult primary brain tumors encountered by oncologists: gliomas, primary central nervous system lymphomas, and meningiomas.
DIAGNOSIS
 CLINICAL FEATURES
CLINICAL FEATURES
Patients with primary brain tumors can present suddenly with seizures or subacutely with progressive focal or non-focal neurological symptoms over several weeks to several months. Progressive focal neurological deficits are usually referable to growth of a tumor in a specific brain location. Non-focal symptoms include headache, vomiting, fatigue, cognitive changes, mood disturbances, imbalance, and gait disorder. Less often, patients may present with an acute stroke-like neurological deficit caused by hemorrhage into a previously subclinical tumor.
Headaches result from local irritation of pain-sensitive dura or from increased intracranial pressure (ICP). Headaches from increased ICP due to tumors are usually holocephalic, progressive, often associated with nausea, worse with recumbency, and may awaken a patient from sleep. They may be precipitated by Valsalva maneuvers or coughing. Systemic symptoms such as malaise, anorexia, weight loss, and fever are usually absent, and presence of these symptoms suggests a metastatic rather than primary brain tumor.
 LABORATORY EVALUATION
LABORATORY EVALUATION
Primary brain tumors are not associated with serologic abnormalities, and no widely accepted primary brain tumor-specific systemic marker exists. Lumbar puncture (LP) for cerebrospinal fluid (CSF) analysis is indicated if there is suspicion of CNS metastasis of systemic cancers or leptomeningeal spread of astrocytoma and for staging of certain primary brain tumors that commonly disseminate in the CSF compartment such as primary CNS lymphoma and primitive neuroectodermal tumor. The CSF may demonstrate an elevated protein level and a mild lymphocytic pleocytosis. A lumbar puncture may precipitate brain herniation if there is increased ICP, and therefore should be performed only after reviewing cranial imaging and obtaining neurological consultation.
 NEUROIMAGING
NEUROIMAGING
The diagnosis of a primary brain tumor is suggested by contrast-enhanced cranial imaging with either computerized tomography (CT) or magnetic resonance imaging (MRI). With the exception of skull bone evaluation, MRI is almost always superior to CT and is the imaging modality of choice.
On CT, tumors and associated vasogenic edema usually appear as regions of low attenuation. Areas of increased attenuation may indicate hemorrhage or calcification. On MRI, tumors often have low signal intensity on T1-weighted sequences and high signal intensity on T2-weighted and FLAIR sequences, although signal characteristics vary with specific tumor types (Figure 62-1). Abnormal enhancement after the administration of intravenous contrast material on either CT or MRI correlates with areas of blood-brain barrier disruption and abnormal endothelial proliferation, and is a surrogate for the location of a tumor. In general, contrast enhancement is characteristic of more malignant tumors.
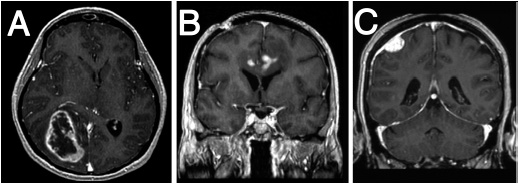
FIGURE 62-1 Magnetic resonance images of primary brain tumors. A. Glioblastoma. Axial T1 contrast-enhanced sequence demonstrating a heterogeneous, peripherally enhancing, centrally necrotic lesion centered in the right temporal-occipital region, causing mass effect on adjacent structures including the corpus callosum and basal ganglia. B. Primary CNS lymphoma. A T1 hypointense mass located in the left cingulate gyrus and subcortical area extending across the corpus callosum and containing several discrete areas of contrast enhancement. C. Meningioma. Coronal T1 contrast-enhanced sequence demonstrates a dural-based, extra-axial mass with intense contrast enhancement and adjacent T1 hypointensity in the brain parenchyma representing vasogenic edema.
Other technologies such as magnetic resonance spectroscopy and positron emission tomography (PET) are useful in specific situations such as surgical guidance and distinguishing between recurrent tumor and treatment-related necrosis. The gold standard for diagnosis, however, remains tissue biopsy.
GLIOMAS
 CLASSIFICATION
CLASSIFICATION
Gliomas account for 80% of all malignant primary brain tumors and 30% of all brain and CNS tumors in adults (2). They consist of cells that display glial differentiation and resemble glial cells (e.g., astrocytes, oligodendrocytes, and ependymal cells). Gliomas comprise a group of tumor types that includes astrocytoma, oligodendroglioma, mixed oligoastrocytoma, ependymoma and other less common variants. Astrocytomas account for approximately 75% of all gliomas and are most often supratentorial, although they may arise anywhere in the CNS.
The WHO categorizes gliomas into four histological grades (1). Grade I is reserved for rare, relatively benign, and slow-growing histological variants including pilocytic astrocytoma, pleomorphic xanthoastrocytoma, and dysembryoplastic neuroepithelial tumor. These tumors carry an excellent prognosis after surgical resection.
Other gliomas are graded using specific histological criteria: cellularity, nuclear atypia, mitotic activity, vascular proliferation, and necrosis. Gliomas that display only one of these features, usually nuclear atypia, are classified as grade II. Gliomas with two features, usually nuclear atypia and mitotic activity, are classified as grade III. Presence of three features, usually nuclear atypia, mitoses, vascular proliferation and/or necrosis, is classified grade IV and these gliomas are termed glioblastoma. Grade II gliomas are commonly referred to as low-grade gliomas (LGGs) while grade III and IV gliomas are commonly referred to as high-grade gliomas (HGGs) or malignant gliomas (MGs).
 LOW-GRADE GLIOMA (LGG)
LOW-GRADE GLIOMA (LGG)
Classification
Most adult low-grade (grade II) gliomas are categorized into three histo-logically defined subgroups: astrocytoma, oligodendroglioma, and oligoastrocytoma. LGGs are relatively uncommon tumors that typically present between the second and fourth decades. They are highly infiltrative tumors that invade and expand contiguous brain tissue.
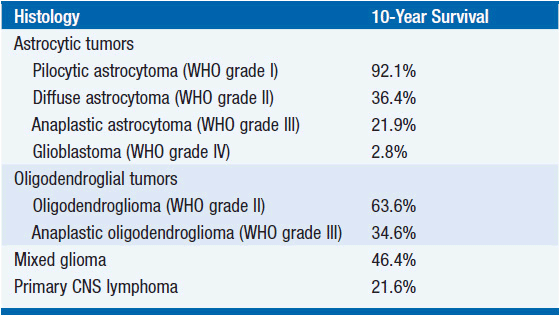
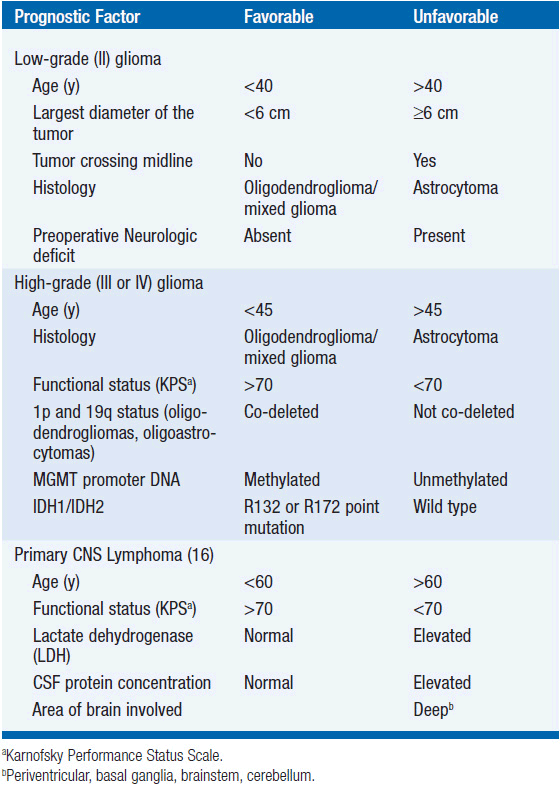
Although initially slow growing, LGGs nearly always progress to HGGs despite treatment. Median survival ranges between 5 and 10 years (Table 62-1) (2). Histological subtype is a critical determinant of survival, with oligodendrogliomas having a more indolent course and better responsiveness to therapy than astrocytomas. Mixed oligoastrocytomas have an intermediate outcome. Other factors that negatively impact survival include age >40 years, the presence of preoperative neurologic deficit, tumor size >6 cm in maximal diameter, and tumor crossing the midline (Table 62-2) (4).
Molecular Profiles of LGG
Proliferation indices such as Ki-67 and MIB-1 are often reported; however, their prognostic relevance in LGG is controversial. Somatic genetic markers have become increasingly useful as potential diagnostic and prognostic tools in LGG. Approximately 80%–90% of low-grade oligodendrogliomas have combined loss of heterozygosity (LOH) on chromosomes 1p and 19q, and loss of both markers correlates with longer survival (5).
Recently discovered alterations in BRAF, IDH1, and IDH2 appear to be hallmark aberrations in particular grade I and II glioma subtypes. Tandem duplication at 7q34 leading to a fusion between KIAA1549 and BRAF results in constitutively active BRAF activity and is found in approximately 70% of pilocytic astrocytomas (6–8). An activating point mutation in BRAF (V600E) is found in an additional 5%–9% of these tumors (9, 10) and in general, RAF alterations occur in ~80% of pilocytic astrocytomas. BRAF V600E mutations are frequently observed (~60%) in other relatively benign glioma variants, including pleomorphic xanthoastrocytoma (10) and ganglioglioma (11), while BRAF tandem duplications are not found in these tumors.
The vast majority (87%) of grade II gliomas (astrocytomas, oligodendrogliomas, and oligoastrocytomas) harbor point mutations in the R132 position of isocitrate dehydrogenase 1 (IDH1) (12) or rarely the analogous codon in IDH2 (R172) (13). Mutant IDH has altered catalytic activity that results in marked accumulation of 2-hydroxyglutarate in gliomas (14), although the functional consequence of this activity is unclear.
Other than 1p/19q co-deletion, the prognostic significance of these somatic alterations remains unclear. However, several have potential diagnostic utility. Pleomorphic xanthoastrocytomas frequently have BRAF V600E mutation, while BRAF tandem duplications (typical of pilocytic astrocytomas) and IDH1/2 mutations (typical of more aggressive grade II gliomas) are rare (10, 13). The vast majority (90%) of IDH mutations in gliomas result in an R132H substitution, which can be detected with a highly sensitive and specific monoclonal antibody. A rapid immunohistochemical analysis using the mutant-specific IDH1 antibody can aid diagnostic analysis (15).
Treatment of LGG
Surgery has an important role in obtaining tissue for histological diagnosis and in providing relief from symptoms caused by mass effect on adjacent structures and increased ICP. No prospective, randomized data exist regarding the benefit of extensive resection of LGGs; however, a number of retrospective analyses suggest cytoreductive surgery improves survival (16). Gross total resection is often advocated for, however, the invasiveness of LGGs and their frequent location near eloquent brain regions often precludes complete excision. Therefore, the primary objective is often maximal safe resection.
Adjuvant treatment options include observation, radiation therapy, and chemotherapy. Radiation is a standard component of treatment for LGGs; however, there remains significant controversy regarding the timing of its implementation. A multicenter, randomized phase III trial demonstrated that early radiotherapy after surgery increases the time to tumor progression but not overall survival compared to radiotherapy at the time of tumor progression (17). However, this study did not stratify for known strong prognostic factors such as oligodendroglial histology and 1p/19q status.
Because patients with LGGs may live for many years, the risk of developing radiation-related adverse effects including cognitive impairment (18) and endocrinopathy (pituitary dysfunction) must be carefully considered. Technical advances in imaging and radiation delivery allow more accurate planning of target volumes and may minimize the long-term side effects of radiotherapy. However, prospective studies incorporating cognitive and quality of life assessments are needed to assess the risk of neurotoxicity and the value of longer time-to-progression with modern radiation techniques.
There is limited but emerging evidence that chemotherapy may be effective in treating LGGs, particularly oligodendrogliomas. Procarbazine, lomustine, and vincristine (PCV) combination therapy and temozolomide have been reported in small series to have activity in both newly diagnosed and progressive LGGs. A randomized phase III trial comparing adjuvant radiotherapy versus temozolomide for newly diagnosed LGG is currently accruing (EORTC 22041).
Because subgroups of LGG patients may survive a long time with little or no neurologic deficit and long-term toxicity is a risk with all therapeutic modalities, management of newly diagnosed LGG remains controversial. Treatment is individualized based on a risk score determined from a number of prognostic factors (Table 62-2) (4). Typically, low-risk patients, particularly those with gross-totally resected tumors, are monitored with serial contrast-enhanced MRI scans. Adjuvant radiation is often provided for high-risk patients, particularly in the setting of incomplete resection.
 MALIGNANT GLIOMAS
MALIGNANT GLIOMAS
Classification and Molecular Profiles of MG
Classification. Malignant gliomas include grade III tumors such as anaplastic astrocytoma, anaplastic oligodendroglioma, and anaplastic oligoas-trocytoma, as well as glioblastoma (GBM, grade IV). GBM accounts for 60% of MGs and is the most common adult malignant primary brain tumor (2). The average age at diagnosis of GBM is 64 years, although it can present at any age.
The prognosis of patients with MG remains unsatisfactory (Table 62-1). The median survival of patients with AA is 2–5 years, while the survival is 10–15 months for those with GBM (2). Prognostic factors associated with improved survival include grade III tumors, oligodendroglial histology, younger age, better performance status, and molecular features such as 1p/19q co-deletion, IDH1/2 mutation (12, 13), and MGMT promoter DNA methylation (19, 20) (Table 62-2). Anaplastic oligodendrogliomas, particularly the 50%–70% with 1p/19q co-deletion, have a better prognosis and are perhaps more responsive to treatment than astrocytomas (21).
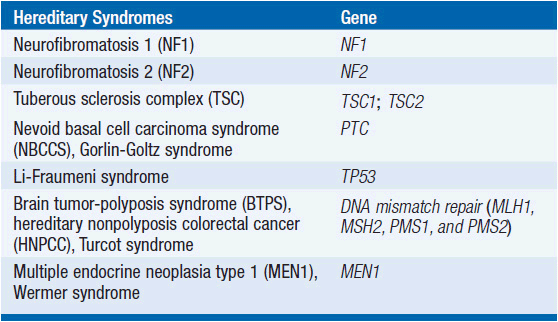
Molecular profiles of GBM. Most GBMs arise as a result of genetic alterations involving tumor suppressor genes and proto-oncogenes within a progenitor cell, although a rare subgroup of gliomas are associated with certain hereditary syndromes (Table 62-3). At least two distinct clinical and genetic forms of GBM have been described (22). Primary GBMs, which account for the vast majority of cases, arise de novo and typically in older patients. These tumors are associated with increased activity of the epidermal growth factor receptor (EGFR), inactivation of the PTEN gene, and normal TP53 status.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


