Unilateral forms of PA (surgically treatable)
Aldosterone-producing adenoma
30–40%
Unilateral adrenal hyperplasia
<5%
Aldosterone-producing carcinoma
Rare
Bilateral forms of PA (medically treated)
Bilateral adrenal hyperplasia
60–70%
Familial hyperaldosteronism type I
Very rare
Pathophysiology of Primary Aldosteronism
Adrenal Pathology
Histological analysis of adrenal glands removed from PA patients indicates that the subdivision of APA and BAH, useful from a clinical and operative perspective, oversimplifies the complex alterations that they undergo: in fact, the adrenals of patients with PA exhibit heterogeneous histological alterations. For example, APAs are rarely composed of a single cell type, but rather, exhibit different cell types in varying proportions comprising: zona fasciculata-like cells, clear cells with large vacuolated lipid-laden cytoplasm, and round central nuclei; lipid-poor zona glomerulosa-like cells; zona reticularis-like cells, compact eosinophilic cells; cells with cytological features of both zona fasciculata (ZF) and zona glomerulosa (ZG) cells showing variable contents of clear lipid micro-vacuoles and granular eosinophilic cytoplasm, also called “hybrid” or “intermediate” cells [13, 14].
Consequently, APA have been classified by some authors according to the prevalent cell type and have attempted to relate the histological findings to some clinical phenotype. For example, the prevalence of ZG-like cells corresponded to responsiveness to angiotensin II infusion and so these APA were denominated, AngII-responsive APA or AR-APA; in contrast to the presence of a prevalence of ZF/reticularis or hybrid cells that correlated to adrenocorticotropic hormone (ACTH) responsiveness (AngII-unresponsive APA, AU-APA) [16]. However, a correlation between cell type and response to posture, and thus to angiotensin II was not observed in another study [17]. Interestingly, most APA patients display either diffuse hyperplasia of the surrounding ZG in the rest of the adrenal or hyperplasia of the ZG with multiple nodules of different sizes, ranging from micro- to macro-nodules [13]. A few reports have studied the contralateral adrenal gland and described similar findings of nodular hyperplasia thereby suggesting that PA may begin, at least in some cases, with one or more unknown stimuli that trigger the development of multiple nodules throughout the adrenal cortex, and a subsequent event stimulates the formation of a single dominant APA that becomes autonomous [14, 15].
In situ hybridization (ISH) studies of CYP11B2 mRNA have demonstrated that in most cases CYP11B2 is overexpressed in the APA; however, in some cases the gene is overexpressed not only in the APA, but also in the surrounding adrenal ZG; whilst in others, CYP11B2 gene expression is not observed in the dominant nodule, but rather, is overexpressed in one or more smaller nodules; in the latter two cases, adrenalectomy did not successfully treat the PA [18, 19].
The recent development of specific antibodies to CYP11B1 (11-beta-hydroxylase) and CYP11B2 (aldosterone synthase) enabled immunohistochemical studies to define the alternative expression patterns of these enzymes from normal adrenals (NA) compared to adrenal glands from PA patients [20].
Normal adrenals display two patterns of expression: (1) a conventional distribution, with CYP11B2 sporadically expressed in the ZG, whereas CYP11B1 is diffusely expressed in the ZF; (2) a variegated pattern with cell clusters strongly expressing CYP11B2 and the remaining areas expressing CYP11B1 [20]. The examination of APA tissues showed that tumors mainly display CYP11B2 positive cells whilst others have a variable proportion of cells expressing the enzyme. APA can consist of three cell types: (a) CYP11B2-positive/CYP11B1-negative cells; (b) CYP11B2-negative/CYP11B1-positive cells; (c) double-negative cells. 3 beta-hydroxysteroid dehydrogenase (3betaHSD) was detected throughout the tumors, irrespective of the expression of CYP11B2 or CYP11B1; whereas CYP17 (17-alpha-hydroxylase) exhibited a similar expression profile to CYP11B1, consistent with its function in cortisol synthesis [20]. In the zone of the adrenal cortex surrounding the APA, two expression patterns have been described: (1) a conventional zonation; and (2) cell clusters expressing CYP11B2 and 3betaHSD, but not CYP11B1, and CYP17, with weak or no expression of CYP11B1 outside the cell clusters [20].
The biochemical and genetic alterations underlying the abnormal cell growth in the zona glomerulosa (ZG) cells of the adrenals of PA patients are still largely unknown. A recent study showed that in rats fed with a high salt diet, although the width of the ZG cells was decreased and the expression of CYP11B2 encoding aldosterone synthase was suppressed, there were nonetheless a number of cells still expressing high levels of CYP11B2 [21]; it is possible to hypothesize that a mutation increased the growth of these cells, thereby resulting in the formation of a nodule of aldosterone-producing cells, not regulated by the increased sodium reabsorption [14].
Genomic Studies in APAs
The molecular basis for the deregulated cell growth and the autonomous hyperproduction of aldosterone observed in PA remains to be fully elucidated. Transcriptosome analysis employing either microarrays or serial analysis of gene expression (SAGE) together with technologies to validate the resultant genetic profiles, such as quantitative real-time PCR (qRT-PCR), have led to several studies attempting to determine the molecular fingerprints of adrenal adenomas. Strict patient selection is of primary importance for such studies and, in fact, insufficiently stringent criteria for APA classification may have caused the discrepancy between the reported differentially expressed genes in APA. Because aldosterone-producing cells are able to store only minimal quantities of the hormone, the synthesis of aldosterone is finely regulated and coupled to its secretion. It is unsurprising therefore, to find aldosterone synthase, up-regulated in APA compared to normal adrenal cortex [22] and all transcriptosome analyses to date have identified the up-regulation of the aldosterone synthase gene (CYP11B2) [23–26]; in contrast, one microarray study identified two sub-groups of APA classified on the basis of a paradoxical up- or down-regulation of a set of gene transcripts that included CYP11B2 [27].
Transcripts that code for other steroid metabolizing enzymes, namely HSD3B2 (encoding 3 beta-hydroxysteroid dehydrogenase type II) and CYP21 (encoding 21-hydroxylase) have also been identified [24]; the latter gene was also found to be overexpressed in APA in a SAGE analysis and validated by ISH [23].
The LHR (luteinizing hormone receptor gene) transcript has been identified as up-regulated by microarray analysis [25, 28] and exogenous expression of the receptor in the aldosterone-secreting adrenocortical carcinoma cell line, NCI H295R [29], and subsequent stimulation with LH resulted in an increased transcription of a luciferase gene reporter construct harboring the CYP11B2 gene promoter.
A second G protein coupled receptor gene, the serotonin receptor 4 (hydroxytryptamine receptor 4, HTR4), that stimulates cAMP release in response to serotonin, has been identified as up-regulated in APA [26, 30, 31]. The aberrant expression of G protein coupled receptors such as LHR and HTR4, among others has been proposed as a putative mechanism for the deregulated steroid production in APA [25, 28].
A recent genomic analysis study by Williams et al. [26] used adenoma tissues from a homogeneously selected group of patients following rigorous diagnostic procedures, including strict criteria for adrenal venous sampling (AVS) interpretation and postadrenalectomy evaluation. Microarray analysis and validation by qRT-PCR identified the epidermal growth factor-like teratocarcinoma-derived growth factor-1 (TDGF1) as the most up-regulated gene in APA compared with NA (21.4-fold). The functional role of TDGF1 was also studied using the H295R cell model: the exogenously expressed growth factor resulted in the activation of phosphatidylinositol 3-kinase (PI3Kinase)/Akt signaling and mediated both an increase in aldosterone secretion (3.8-fold) as well as an inhibition of apoptosis. Both functional effects were blocked by PI3Kinase inhibitors [26].
Interestingly, a sixfold up-regulation of the TDGF1 gene transcript in APA had been identified previously in another study by SAGE [23]. Therefore, two independent methods of transcriptosome analysis have identified TDGF1 as an up-regulated gene transcript in APA compared to normal tissue [23, 26] that, considered together with the functional data, indicate that this gene may represent a key player in the development and pathophysiology of PA.
Role of Aldosterone in Target Organ Damage
Traditionally, aldosterone has been considered the main regulator of water and electrolyte homeostasis due to its effects on epithelial cells, particularly in the collecting ducts of the kidney and distal colon. The physiological actions of aldosterone in non-epithelial tissues are much less evident, but over the last 15 years a wealth of studies both in humans and animal models have allowed new insights on its pathophysiological effects, that are mainly targeted in the heart, blood vessels, kidney, and central nervous system (CNS).
Heart
The main pathological effects of aldosterone excess on the heart are vascular and perivascular inflammation, fibrosis, and myocardial hypertrophy. In a study by Rocha et al. [32] administration of aldosterone and a salt-rich diet to uni-nephrectomized rats resulted in severe arterial hypertension and inflammatory lesions in myocardial vessels, with fibrinoid necrosis in the media and mononuclear leukocyte accumulation in perivascular tissues. These changes were prevented by concomitant administration of the mineralocorticoid receptor antagonist eplerenone, an effect that was independent from blood pressure reduction; similar results have been achieved in other studies [33]. A direct role of aldosterone in myocardial inflammatory damage was further demonstrated in transgenic rats overexpressing the human renin and angiotensinogen genes (dTGR); the hypersecretion of aldosterone promoted hypertrophy, cardiac remodeling and fibrosis, independent of blood pressure. These effects involved an increased expression of NF-kappaB, a key transcription-factor involved in the inflammatory response, and the basic fibroblast growth factor (bFGF) [34]. Therefore, myocardial fibrosis can be determined both as a direct effect of stimulation of cardiac fibroblast proliferation as well as an indirect reparative response to inflammation and cell death [12].
Clinical studies suggested that PA is associated with increased prevalence and severity of left ventricular hypertrophy (LVH) [35]. Napoli et al. [36] demonstrated that PA patients displayed a significantly higher severity of LVH after correction for age, gender, duration, and severity of hypertension compared to essential hypertensives. Myocardial perfusion evaluated by myocardial scintigraphy is significantly more impaired in PA patients compared with matched hypertensive subjects [36]. Rossi et al. [35] demonstrated that, in essential hypertensive and in PA patients, plasma aldosterone levels were directly related to LV wall thickness. Further, myocardial texture was also modified by plasma aldosterone levels [35–37], with an increase in the fibrotic components, resulting in diastolic dysfunction.
In addition to these effects, aldosterone is able to directly affect membrane ionic balance in cardiomyocytes, acting on ionic channels and transporters. Aldosterone reduces Na+-K+-pump affinity for sodium and potassium [38], causing a disequilibrium in intra- and extracellular concentrations of these cations, and modifies potassium fluxes during repolarization, especially acting on the rapid component of the delayed rectifier K+-current. These changes causes a lengthening of the QT-interval at ECG, and can be reversed by spironolactone [39]. Aldosterone also acts on the Na2+/Mg2+-exchanger, with a resultant depletion in intracellular magnesium concentration, and stimulates intracellular calcium influx [40] causing calcium intracellular excess, which can elicit ectopic ventricular beats [41].
Blood Vessels
Vascular and perivascular inflammation elicited by aldosterone excess in myocardium can also be observed in other vascular beds, with similar mechanisms of endothelial activation, leukocyte accumulation, and pro-fibrogenic cytokine production. Aldosterone is in fact able to determine the loss of physiological vasodilating properties of endothelial cells and their imbalance towards a pro-thrombotic and pro-inflammatory phenotype known as “endothelial dysfunction” [42] and also an increased vascular stiffness. Some studies have investigated the mechanisms responsible for these changes, such as overexpression of cyclooxygenase type 2 (COX-2) and overproduction of prostanoids [42], overproduction of endothelin-1 (ET-1) [43] and decrease in expression and activity of NO-synthase [44] with reduced levels of nitric oxide (NO), a well-known vasodilating factor. In agreement with the increased vascular deposition of collagen, a higher level of fibrosis in the walls of subcutaneous small resistance arteries of PA patients has been demonstrated [45].
Kidney
Many studies with animal models of mineralocorticoid excess have shown the involvement of aldosterone in the pathogenesis of renal disease. The first complete demonstration of this causal association was given by the so-called remnant kidney model. In 1996, Greene et al. [46] submitted normotensive rats to sub-total nephrectomy and then compared a group treated with both an ACE-inhibitor (enalapril) and an angiotensin II receptor antagonist (losartan), a group treated with enalapril, losartan, and exogenous aldosterone and a control group without any treatment. Animals in the control group developed arterial hypertension, proteinuria, and glomerulosclerosis associated with overactivity of the renin–angiotensin–aldosterone system (RAAS). The renal lesions that developed in rats treated with RAAS-inhibitors only, were significantly less severe; this protective effect, however, was lost with concomitant administration of exogenous aldosterone. Interestingly, this demonstrate not only a role for aldosterone in renal disease but distinguished its effects from those of angiotensin II. Other animal models were subsequently investigated, for example, spontaneously hypertensive rats—stroke prone (SHRSP), a model of secondary aldosteronism, prematurely develop proteinuria, and malignant nephrosclerosis on a salt-rich diet [47, 48]. Adrenalectomy [33] or mineralocorticoid receptor blocker therapy [33, 48] has proven effective in preventing renal changes in a blood pressure-independent manner [48]. In a model of aldosterone/salt-treated rats, the animals develop severe hypertension and albuminuria, renal vascular damage, and renal lesions together with myocardial and coronary vascular damage, compared to rats receiving salt alone. Eplerenone significantly reduced blood pressure levels, albuminuria, and renal vascular injury. Renal vascular inflammation after 4 weeks of aldosterone administration was demonstrated by showing elevated expression of osteopontin, monocyte chemoattractant protein, interleukin-1, and interleukin-6. Treatment with eplerenone resulted in renal vascular, glomerular and tubule protection, and attenuated vascular inflammation [49].
Central Nervous System
Mineralocorticoid receptors have been located in several regions of the CNS, especially in peri-ventricular areas, amygdalae, hypothalamus, and cerebellum [50]. The CNS is now considered one of the main targets of aldosterone, as this hormone has been associated with modulation of ACTH secretion, maintenance of arousal and central modulation of salt appetite, water and electrolyte homeostasis, and blood pressure levels [51, 52]. Raised aldosterone levels in CNS can then increase peripheral blood pressure even in the presence of normal plasma aldosterone concentrations [53, 54]. The main mechanisms involved in this effect seem to be the increase in salt appetite, the expansion in extracellular fluid volume, and a marked sympathetic activity [12]; moreover, some studies carried out in dogs have observed a reduction in baroreflex sensitivity following administration of exogenous aldosterone both in acute and in chronic conditions [55].
The main consequence of aldosterone excess on the CNS is the predisposition to acute cerebrovascular events, related to vascular damage that aldosterone may induce in the CNS vascular bed. SHRSP represented a key model to study the causal association between aldosterone and stroke. These animals develop malignant hypertension by 7–10 weeks on salt-rich diet depleted in potassium, with a paradoxical activation of the RAAS and onset of nephroangiosclerosis and premature ischemic stroke which usually results in death within 13–18 weeks [32]. Treatment with mineralocorticoid-receptor blockers can prevent development of severe vascular lesions and decrease mortality in a pressure-independent manner [56].
Clinical Features and Complications of PA
The most relevant feature of primary aldosteronism is an excessive and inappropriate secretion of aldosterone, in relation to blood volume and pressure and to plasma sodium levels. The subsequent rise in blood pressure determines suppression of renin secretion and decrease of angiotensin II production. The excess in aldosterone induces potassium secretion and sodium reabsorption by kidneys, and therefore an electrolyte imbalance; this also determines an increase in magnesium and hydrogen ion excretion, which induces the retention of bicarbonate ions and consequently the onset of metabolic alkalosis. However, even with the powerful effects of aldosterone as kaliuretic and sodium retention agent, plasma concentrations of sodium and potassium can still be within the normal range, due to the activation of compensatory mechanisms.
Sodium retention, in particular, is compensated by escape natriuretic mechanisms, such as an increase in renal perfusion pressure, a rise in natriuretic peptides, and a decrease in sympathetic nervous renal activity and in plasma concentration of angiotensin II [57]; it is because of these mechanisms that patients with primary aldosteronism do not show oedemas.
Hyperaldosteronism induces an increase in blood pressure by sodium retention and the subsequent increase of circulatory volume and cardiac output, which is normalized by reflex vasoconstriction; ultimately, this determines the onset of hypertension. Moreover, recent evidence suggests a direct vasoconstrictory activity of aldosterone itself, in accordance with its rapid secretion after postural changes [58].
Paresthesias and neuromuscular disorders are relatively common in Asian countries [59], but the symptoms are often non specific; in fact, hypokalemia affects only of a relatively small percentage of subjects (less than 30%), who describe muscular weakness and cramps, polydipsia, polyuria, or nycturia.
The two most common features are hypertension and hypokalemia. Hypertension can be moderate or severe, and is often resistant to common antihypertensive therapy; however PA patients with normotension have been nonetheless reported.
A marked hypokalemia can be associated to alterations of electric conduction in myocardium, which manifests as EKG changes, such as T wave flattening, onset of U wave, ST segment sub-elevation, prolongation of PQ, and QT intervals and of QRS; all these conditions can lead to potentially fatal ventricular arrhythmias. It has been shown that PA patients display an increased prevalence of long QT, at least in part independent from potassium levels [60], which is reverted after adrenalectomy or therapy with spironolactone [61].
In the latest decade, many studies have compared patients with PA and essential hypertension, confirming results in animal models that indicate aldosterone per se increases cardiovascular risk, independent of the effect on blood pressure levels.
Patients with PA display a higher incidence of metabolic syndrome compared to essential hypertension [62], and even those without this condition often show a more pronounced insulin-resistance [62, 63]. Metabolic alterations, in turn, can be in part responsible for the increased cardiovascular risk of PA patients [64]. These alterations are ameliorated by PA treatment by spironolactone or adrenalectomy [65]. To date it has not been clarified if metabolic syndrome is secondary to hyperaldosteronism or vice versa. In fact, it has been shown that adipokines such as complement-C1q TNF-related protein 1 (CTRP1), stimulates aldosterone production through an increased expression of CYP11B2 [66]. In the other hand, a longitudinal evaluation of the incidence of metabolic syndrome and changes in metabolic risk factors in the Framingham Offspring Study, showed that higher quartiles of aldosterone were associated to a higher risk of development of metabolic syndrome [67]. However, another study was not able to confirm the association between metabolic syndrome and PA [68]. Finally, a recent retrospective study from the German Conn’s Registry, showed that PA patients displayed a higher rate of diabetes compared to a control population of essential hypertensives [69] and others reported a high prevalence of PA in type 2 diabetic patients with resistant hypertension [70, 71].
PA patients also show a significantly higher rate of structural and functional alterations of the heart, kidneys, and blood vessels if compared to patients with essential hypertension. PA patients showed a higher prevalence and degree of LVH compared to essential hypertensives [11, 35, 62] and also a higher prevalence of diastolic dysfunction [72]. At vascular level, PA patients show an increased intima-media thickness [73] and an increased arterial wall stiffness, measured with the pulse wave velocity [74].
PA patients also show a high prevalence of obstructive sleep apnea (OSA) [75]. Calhoun et al. [76] observed that subjects at high risk for OSA were almost two times more likely to have PA diagnosed and had a higher urinary aldosterone excretion. Subsequently, they showed a significant correlation between plasma aldosterone concentration and the severity of OSA [77]. The link between these two conditions is not straightforward: obesity is a common feature in OSA patients and thus, cytokines produced by the adipose tissue may stimulate aldosterone secretion from the adrenals; on the other hand, aldosterone-induced fluid retention may contribute to the upper airway obstruction via an increase of the peripharyngeal oedema.
Few studies have compared renal damage in PA and essential hypertension; the former, though, has higher levels of albuminuria and a higher prevalence of microabuminuria [78–80].
The difference is evident after adrenalectomy or spironolactone therapy; it has been therefore suggested that an increase in albumin urinary excretion could be an indicator of functional damage in early renal disease. Sechi et al. have observed, through the use of Doppler ultrasonography, reduced renal vascular resistances in PA patients when compared to those of essential hypertension; moreover, glomerular filtration rate (GFR) based on creatinine clearance is higher in PA patients [81]. In subjects with higher clearance levels than the population median, the hemodynamic alterations were reversible after adrenalectomy or medical treatment with mineralocorticoid receptor antagonists. Moreover, even though renal excretion of albumin was higher in PA patients than in essential hypertensives, urinary albumin/creatinin ratio (AU/CU) did not show a significant difference. These data, as a whole, confirm the presence of glomerular hyperfiltration in PA, as previously reported [78, 80], and correlate these finding to anomalies in intra-renal vascular resistances, which appear responsible for the elevated albumin excretion. However, long-term exposure to high aldosterone levels results in a deterioration of renal function. In the German Conn’s Registry, the percentage of patients with a serum creatinine concentration above the normal range was higher in PA patients than in hypertensive controls: age, male sex, low potassium, and high aldosterone concentrations were independent predictors of a lower GFR [82].
Considering the elevated prevalence of multiple cardiovascular risk factors and increased target organ damage in PA patients, an increased prevalence of cardio- and cerebrovascular events is also expected; in fact, PA patients display a fourfold increase in the prevalence of stroke, 6.5-fold of myocardial infarction and 12-fold of atrial fibrillation [11]. These data have been confirmed by further studies [75, 83]. Interestingly, in one of these studies, normokalemic PA patients displayed a higher risk of stroke, highlighting the importance to exclude PA not only in hypokalemic, but also in normokalemic hypertensives [75].
Diagnosis of Primary Aldosteronism
Screening Test
The Guidelines for the diagnosis and treatment of PA recently published the different categories of hypertensive patients that display a relatively high prevalence of PA; it is recommended that patients belonging to these categories undergo a screening test. The screening test should be performed in all patients with: (1) resistant hypertension; (2) hypertension grade 2 or 3; (3) hypertension and spontaneous or diuretic-induced hypokalemia; (4) hypertension with adrenal incidentaloma; (5) hypertension and a family history of early-onset hypertension or cerebrovascular accident at a young age (<40 year); (6) all hypertensive first-degree relatives of patients with PA [4] (Fig. 1.1).
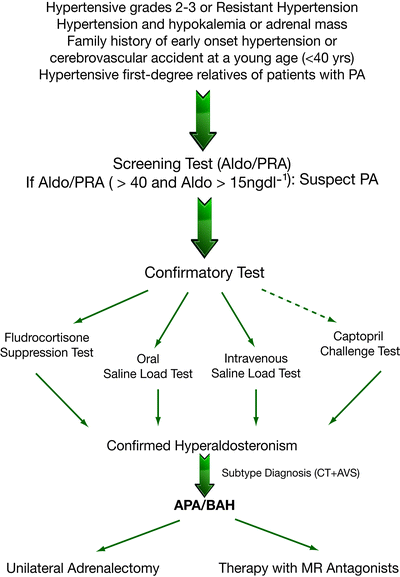
Fig. 1.1
Flow-chart for diagnosis of PA and its subtypes
The aldosterone to renin ratio (ARR) is the most reliable, currently available method for PA screening: in fact, many studies have demonstrated that the ARR is superior to measurements of both potassium and aldosterone (which are less sensitive) as well as renin alone (which is less specific) [84–86]. Presently, there is no general consensus on ARR cut-off and therefore, individual centers use different values, ranging from 7.2 to 100 ng dL−1 per ng mL−1 h−1, with a consequent wide variation in the sensitivity (64–100%) and specificity (87–100%) of the screening test. However, in a recent study published by Rossi et al., the ARR was found to provide a good within-patient reproducibility and an accuracy of 80% for identifying APA patients [87]. The most important confounding factors affecting renin and/or aldosterone measurements thus reducing the sensitivity or specificity of the ARR are antihypertensive drugs [4] (Fig. 1.2). In particular, β-blockers, α-methyldopa and clonidine decrease the beta-adrenoreceptor mediated stimulation of renin production and sympathetic nervous system output thereby causing false-positives. Conversely, other drugs stimulate renin secretion, such as ACE-inhibitors, ATII receptor antagonists, dihydropyridine calcium antagonists, and diuretics. All these drugs (except diuretics that increase aldosterone secretion) may cause a reduction in aldosterone levels and increase in renin levels leading to false-negative results. The recently introduced renin inhibitor Aliskiren reduces plasma aldosterone levels whilst stimulating renin secretion: this results in false-positive ARR levels for renin measured as plasma renin activity (PRA) and false negatives for renin measured as direct renin concentration (DRC) [4]. Antihypertensive medications other than diuretics, that should always be withdrawn for at least 4–6 weeks (6–8 weeks for spironolactone), should be stopped at least 2–3 weeks before ARR testing, but in many patients this is clearly not feasible. Because α-antagonists do not appear to have any significant effect on plasma aldosterone and renin levels, they can be safely used to control hypertension before the screening test is performed. Non dihydropyridine calcium antagonists can cause a modest increase in renin levels and reduce aldosterone secretion, but rarely to an extent that can significantly affect the ARR and confirmatory tests and can be administered to patients whose blood pressure is not adequately controlled by an α-antagonist alone [4, 88].
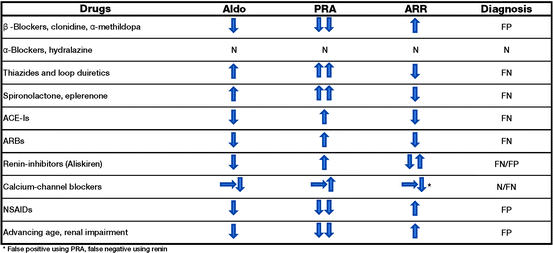
Fig. 1.2
Effects of drugs on aldosterone, plasma renin activity (PRA), aldosterone to renin ratio (ARR) levels and screening test
Further, the effect of oral contraceptive agents has received little attention, but should be considered because estrogen-containing preparations induce angiotensinogen synthesis by the liver. Blood sampling conditions can also have an effect and should be standardized to avoid potential fluctuations in levels of the two hormones and subsequent difficulties in interpreting the ARR [4] (Table 1.2).
Table 1.2
Measurement of aldosterone:renin ratio (ARR)
ARR measurement |
|---|
Preparation for ARR measurement |
Correct hypokalemia |
Avoid sodium restriction |
Withdraw agents that affect the ARR (see Fig. 1.1) |
Conditions for blood collection |
Collect blood mid-morning, after the patient has been up for at least 2 h and seated for 5–15 min |
Collect blood avoiding stasis and hemolysis |
Maintain blood sample at room temperature and not on ice (avoiding activation of renin) |
Separate plasma from cells within 30 min of collection |
Factors to take into account when interpreting the results |
Age |
Medications |
Method of blood collection |
Level of potassium and creatinine |
The absolute aldosterone value (>15 ng dL−1) and the lowest detectable level of PRA should also be taken into account. Because the ARR is dependent on PRA, effectively anyone with suppressed PRA will have an increased ARR; this is particularly important in elderly or black populations, who often display PRA values as low as 0.1 ng mL−1 h−1, thereby resulting in an increase in the plasma aldosterone concentration: plasma renin activity (PAC:PRA) ratio even with a PAC of 5 ng dL−1. It has been shown that all individuals with aldosterone levels of less than 9 ng dL−1 demonstrate normal suppression during a fludrocortisone-suppression test [89].
In the majority of published studies, ARR is reported as a function of renin, measured as PRA, and few data are available for direct active renin (DAR). PRA and DAR are closely correlated (overall correlation coefficient 0.98), but the correlation is weaker for the low compared to the high/normal range of values (r = 0.77 for samples with PRA <0.65 ng mL−1 h−1), i.e., in the range of patients potentially affected by PA [90, 91]. Even newer automated methods for renin measurements have not resolved this issue (r = 0.14 with PRA < 1) [92]. To date, no prospective studies have been performed that have compared the accuracy of the aldosterone to active renin ratio to that of the well-established ARR and therefore, DAR should be considered cautiously as a screening test for PA.
In light of the high prevalence of low renin hypertension [90], it is important to emphasize that an increased ARR is not itself diagnostic of PA and a confirmatory test is always required to avoid patients unnecessarily undergoing costly and potentially harmful lateralization procedures.
Confirmatory Tests
Although the Guidelines clearly recommend that patients with a positive ARR undergo a confirmatory test to definitively confirm or exclude the diagnosis of PA, the choice of test remains a matter of debate and there is currently insufficient direct evidence to recommend one rather than another [4] (Fig. 1.1).
Four testing procedures are approved by the Guidelines: oral sodium loading (OLT), saline infusion (SLT), fludrocortisone suppression (FST), and a captopril challenge (CCT) [4] (Table 1.3).
Table 1.3
Confirmatory tests for PA
Primary aldosteronism confirmatory tests | ||
|---|---|---|
Test | Procedure | Interpretation |
Oral sodium loading test—OLT | Oral NaCl supplementation (>200 mmol Na+/day for 3 days) and K+ supplementation to maintain plasma K+ in normal range | PA unlikely if urinary Aldo <10 μg/24 h in the absence of renal disease. Elevated urinary Aldo excretion >12 μg/24 h indicates PA highly likely |
Urinary Aldo measured in 24 h urine collection from morning of day 3 to morning of day 4 | ||
Intravenous saline loading test—SLT | Intravenous infusion of 2 L 0.9% NaCl solution over 4 h (500 mL h−1) with patient remaining in recumbent position | Post-infusion plasma Aldo levels <5 ng dL−1 indicate no PA; values >10 ng dL−1 indicate PA; values between 5 and 10 ng dL−1 are inconclusive (57–60); most units consider these patients as PA |
Blood samples for renin and Aldo measurements collected at time zero and after 4 h, with blood pressure and heart rate monitored during the test | ||
Fludrocortisone suppression test—FST | Fludrocortisone acetate (0.1 mg every 6 h), K+ supplementation (every 6 h at doses sufficient to maintain plasma K+, measured four times a day, close to 4 mmol L−1) and NaCl (30 mmol every 8 h) and dietary salt to maintain urinary Na+ excretion rate of at least 3 mmol kg−1 body weight for 4 days. On day 4, plasma Aldo and PRA are measured at 10 h with patient seated, and plasma cortisol is measured at 7 and 10 h | Upright plasma Aldo >6 ng dL−1 on day 4 at 10 h confirms PA, provided PRA is <1 ng mL−1 h−1 and plasma cortisol concentration is lower than the value obtained at 7 h (to exclude a confounding ACTH effect) |
Captopril challenge test—CCT | 25–50 mg captopril orally after sitting or standing for at least 1 h. Blood samples are collected for PRA measurement, plasma Aldo at time zero and at 1 or 2 h after captopril challenge, with patient remaining seated during this period | In patients with PA, aldosterone remains elevated and PRA remain suppressed |
PA is confirmed when the post-captopril ARR is >30–40 or post-captopril Aldo is >8.5–10 ng dL−1 | ||
Briefly, FST consists of fludrocortisone acetate administration for 4 days with KCl and NaCl supplements. If PAC on the fourth day is ≥6 ng dL−1, PA is confirmed (Table 1.3) [4, 16, 93].
Related posts:
 Congenital Adrenal Hyperplasia
Congenital Adrenal Hyperplasia
 Central Mineralocorticoid Receptors and Cardiovascular Disease
Insulin Resistance and Hypertension
Central Mineralocorticoid Receptors and Cardiovascular Disease
Insulin Resistance and Hypertension
 Syndromes of Mineralocorticoid Excess
Syndromes of Mineralocorticoid Excess
 Testosterone Deficiency or Male Hypogonadism
Testosterone Deficiency or Male Hypogonadism
 Primary Generalized Familial and Sporadic Glucocorticoid Resistance (Chrousos Syndrome) and Hypersensitivity
Primary Generalized Familial and Sporadic Glucocorticoid Resistance (Chrousos Syndrome) and Hypersensitivity
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


