Abstract
Polycystic ovary syndrome is the most common endocrinopathy of reproductive-aged women. Major clinical manifestations include hirsutism and irregular menstrual bleeding as a result of altered hypothalamic-pituitary-ovarian interaction. Metabolic dysfunction is common and includes insulin resistance, lipid abnormalities, and obesity. There is increased risk for diabetes, uterine cancer, and cardiovascular disease.
Keywords
Hyperandrogenemia, anovulation, polycystic ovary, insulin resistance, obesity, puberty, folliculogenesis, granulosa, theca
Understanding the clinical significance and pathophysiology of polycystic ovary syndrome (PCOS) has evolved over more than 150 years. The first description of enlarged, polycystic ovaries surrounded by a smooth capsule was reported in 1844. This was followed by similar observations, including a description on hyperthecosis in 1897. In the early 1900s, there was a growing awareness that dysfunctional uterine bleeding was associated with multiple cystic follicles of the ovary, which, in part, led to the therapeutic recommendation of bilateral ovarian wedge resection. In 1926, it was demonstrated in rodents that gonadotropic extract derived from the urine of pregnant women was capable of inducing multiple ovarian cyst formations. This finding suggested that abnormal secretion of anterior pituitary hormones may be responsible for the morphological changes in the ovary. Subsequently, in 1935, the classic description of polycystic ovaries was reported by Stein and Leventhal, which codified the association with hyperandrogenism, amenorrhea, and infertility as well as established the syndrome named for the authors. As investigators began to study the pathogenesis of this disorder and the number of relevant publications increased, there followed a gradual and distinct terminological conversion to what has become known as PCOS.
Currently the term PCOS refers to a multisystem reproductive-metabolic disorder that has evolved over decades and stands to be further defined in years to come. The principal clinical manifestations are hyperandrogenism and irregular menstruation, the latter of which leads to infertility. The ovaries of women with these symptoms are polycystic and conform to a specific anatomical appearance that may be detected on ultrasound imaging, although the ovarian morphology can exist in women without the overt clinical manifestations. The associated metabolic dysfunction includes insulin resistance, dyslipidemia, and, in the United States, an increasing prevalence of obesity. The intriguing nature of PCOS rests with the underlying mechanism(s) responsible for the juxtaposed abnormalities of hypothalamic-pituitary-ovarian-adrenal function with those of altered metabolic physiology. All of these factors may contribute to the clinical phenotype, pose increased long-term health risks, and serve as targets for therapeutic intervention in women with PCOS.
Diagnostic Criteria
- ◆
The central components of PCOS are hyperandrogenism, oligo-anovulation, and polycystic ovarian morphology, excluding other endocrinopathies.
- ◆
Different PCOS phenotypes exist by Androgen Excess and PCOS (AE-PCOS) and Rotterdam criteria and vary in their degree of reproductive and metabolic dysfunction.
- ◆
The combination of hyperandrogenism with menstrual irregularity increases the risk of metabolic dysfunction.
The 1990 National Institutes of Health (NIH)-sponsored conference defined PCOS as hyperandrogenism and/or hyperandrogenemia with oligo-anovulation, excluding other endocrinopathies, including congenital adrenal hyperplasia (CAH), Cushing syndrome, thyroid dysfunction, hyperprolactinemia, androgen-producing tumors, and drug-induced androgen excess ( Table 21.1 ). Several years later, the Rotterdam consensus modified the diagnostic criteria to include at least two of the following three features: (1) clinical or biochemical hyperandrogenism, (2) oligo-anovulation, and (3) polycystic ovaries, excluding the previously described endocrinopathies. These newer Rotterdam criteria for PCOS are currently recommended for clinical use and include all patients defined by 1990 NIH criteria (i.e., classic PCOS) along with women with either (1) clinical/biochemical hyperandrogenism and polycystic ovaries (i.e., ovulatory PCOS) or (2) polycystic ovaries with ovulatory dysfunction. Consequently, the 6% to 10% prevalence of PCOS by 1990 NIH criteria has doubled with the use of the broader Rotterdam criteria, with 1990 NIH-defined PCOS being the most common phenotype.
| 1990 National Institutes of Health Criteria (Both 1 and 2) |
|
| Revised 2003 Rotterdam Criteria (At Least 2 Out of 3) |
|
| Androgen Excess-polycystic ovary syndrome (AE-PCOS) Society Criteria (Both 1 and 2) |
|
* Excluding other endocrinopathies, including congenital adrenal hyperplasia, Cushing syndrome, thyroid dysfunction, hyperprolactinemia, androgen-producing tumors, and drug-induced androgen excess.
The AE-PCOS Society criteria for the diagnosis of PCOS represent a modification of the 1990 NIH/National Institute of Child Health and Human Development (NICHD) criteria by considering hyperandrogenism as the cardinal feature of PCOS. Consequently, the AE-PCOS defines PCOS as (1) hyperandrogenism (clinical and/or biochemical) and (2) ovarian dysfunction (oligo-anovulation and/or polycystic ovaries), (3) excluding other androgen excess-related disorders. Using AE-PCOS criteria for PCOS, patients with clinical/biochemical hyperandrogenism and polycystic ovaries who are ovulatory would be considered to have PCOS (i.e., ovulatory PCOS by Rotterdam criteria), while nonhyperandrogenic patients with polycystic ovaries and ovulatory dysfunction would not (i.e., polycystic ovaries with ovulatory dysfunction by Rotterdam criteria).
The importance of categorizing PCOS by different phenotypes is that women with PCOS, as defined by NIH criteria, are at increased risk of developing reproductive and metabolic abnormalities, including menstrual dysfunction, anovulatory infertility, type 2 diabetes mellitus, and metabolic syndrome, respectively. In contrast, ovulatory PCOS patients, included in Rotterdam and AE-PCOS criteria, have a lower body mass index (BMI) and lesser degrees of hyperinsulinemia and hyperandrogenism than women with NIH-defined PCOS, which may contribute to lowered risks of developing similar reproductive and metabolic abnormalities. Women with combined polycystic ovaries and oligo-anovulation (without androgen excess) appear least affected with respect to metabolic risk.
Epidemiology
- ◆
PCOS is the most common endocrinopathy of reproductive-aged women.
- ◆
Female offspring of women with PCOS are at increased risk for the syndrome.
Prevalence
According to NIH criteria for PCOS, chronic anovulation, and clinical/biochemical evidence of androgen excess, the estimated prevalence of this disorder has been reported to range from 4% to 10% of women in their reproductive years, which designates PCOS as the most common endocrinopathy of women. In a study of 277 unselected women undergoing employee entrance physical examinations, 4.6% were found to have PCOS. This rate is consistent with a reported 8.0% prevalence of PCOS among 230 young women with irregular menstrual bleeding and hirsutism by Ferriman-Gallway scoring. These results confirmed earlier reports in which the diagnosis of PCOS was highly likely in women with hirsutism and oligomenorrhea.
The subsequent introduction of expanded criteria by the Rotterdam Consensus Conference and the AE-PCOS Society to include ovarian morphology by ultrasonography has increased the prevalence of this disorder. Using Rotterdam criteria, a prevalence of 19.9% was detected in approximately 400 women of Turkish descent compared with 6.1% as defined by NIH criteria. In an indigenous population of 248 Aboriginal and/or Torres Strait Islanders, the prevalence of PCOS was 20.9% as defined by Rotterdam and 15.3% according to NIH criteria. Similar to that of Rotterdam, diagnostic criteria for PCOS established by the AE-PCOS Society also led to an increased prevalence, 15.3%, in Turkish women.
In the absence of hirsutism, it has been demonstrated that women with anovulation have a high incidence of hyperandrogenemia, conferring a diagnosis of PCOS. In 87 women presenting with oligomenorrhea, 87% exhibited elevated levels of circulating testosterone (T) while in those with amenorrhea, 32% were noted to be hyperandrogenemic. In 206 nonhirsute women, biochemical assessment revealed elevated androgens consistent with the diagnosis in 87% of those with oligomenorrhea and 32% with amenorrhea. In contrast, most women with hirsutism alone were reported to have ultrasound findings of polycystic ovaries regardless of whether the patient had oligomenorrhea, amenorrhea, or regular cycles. In these women, increased circulating androgen levels may have facilitated the development of polycystic ovaries. This notion is supported by studies in which female to male transsexuals treated with androgen acquire the typical appearance of polycystic ovaries.
Familial Occurrence
It has been well documented that PCOS tends to aggregate within families. Moreover, several studies have addressed the prevalence of PCOS among first-degree relatives. Based on clinical evidence and laboratory confirmation of hyperandrogenism, anovulation, and polycystic ovaries, the likelihood of PCOS in sisters and mothers of affected women has been reported to be considerably higher than those of normal controls. In a study of 115 sisters of 80 probands, PCOS was demonstrated in 22% of reproductive-aged siblings, whereas hyperandrogenemia was found in an additional 24%. Additional studies of families with PCOS have revealed that 32% to 66% of sisters and 24% to 52% of mothers had the syndrome. In contrast, a study of sisters and mothers of Turkish women with PCOS revealed rates of 16% and 8%, respectively. In most of these studies, the prevalence of insulin resistance and impaired glucose tolerance was considerably greater in the relatives compared with normal controls.
A PCOS phenotype for men and male siblings of affected women with PCOS has not been established. However, circulating dehydroepiandrosterone sulfate (DHEA-S) levels in first-degree male relatives have been shown to be significantly higher than those of unrelated BMI-matched controls. Serum T, sex hormone binding globulin (SHBG), luteinizing hormone (LH), and follicle-stimulating hormone (FSH) were similar between groups.
Collectively, these results indicate that first-degree relatives of PCOS women are at significant risk for having PCOS. The findings support a genetic basis for hyperandrogenemia, which may account, at least in part, for the familial clustering of this disorder .
Clinical Description
- ◆
The onset of excess hair growth occurs during or soon after puberty.
- ◆
Irregular menstrual bleeding commonly persists well after menarche.
- ◆
The polycystic ovary is characteristic of, but not unique to, PCOS.
- ◆
Obesity and abnormal insulin secretion are present in most women with PCOS.
- ◆
Women with PCOS frequently seek treatment for infertility.
Hirsutism
The most distinctive and visible clinical feature of PCOS is hirsutism, the degree of which varies from mild to severe. The rate of hair growth is important clinically; gradual and progressive growth indicates a functional etiology, whereas the rapid appearance of thick, pigmented hair often suggests a neoplastic source of androgen production. In PCOS, increased hair growth is commonly found on the side of the face, upper lip, and chin extending down to the neck region ( Fig. 21.1 ). This pattern of hirsutism may be accompanied by extension of pubic hair growth towards the umbilicus and may include excess hair on the extremities, abdominal flank, and back, although these latter areas are not considered specific sites of sexual hair growth. More severe cases include the appearance of hair on the chest. Progressive hyperandrogenism may be associated with temporal balding and male pattern baldness. In PCOS, the amount of hirsutism has been variably correlated to serum androgen concentrations. However, individual variation in hair growth may reflect ethnic differences, which may account for slight variations of prevalence of PCOS in different parts of the world.
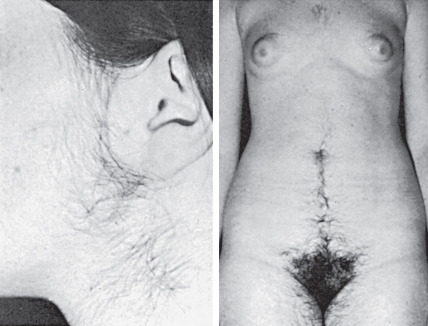
Hirsutism is usually defined by a modified Ferriman-Gallwey (F-G) score of at least eight in black or white women of reproductive age. Limitations of the F-G score include its subjective nature, poor correlation with hyperandrogenemia, failure to account for hair growth in other areas (i.e., sideburns, buttocks), different hirsutism scores in various populations, and inability to assess the psychological impact of hirsutism on a woman’s well-being. With history and exam, however, it can identify selected hirsute women at risk for underlying disease requiring treatment, affecting fertility, or having medical or genetic implications.
There have been several attempts to assess the activity of the androgen receptor (AR) in PCOS women. However, the results have been inconsistent. The AR gene encodes variable length CAG repeat polymorphism in the transaction domain of the X chromosome to determine activity of the AR protein. It has been shown that the normal CAG number ranges from 8 to 35. Notably, length of the CAG repeat and AR activity are inversely correlated. In a recent large study of 330 women with PCOS and 289 controls, shorter CAG repeat number was associated with PCOS women. The distinguishing feature of this study was that NIH criteria were used for the diagnosis of PCOS, which ensured evidence of hyperandrogenemia, whereas most prior studies used criteria that did not necessarily include hyperandrogenism. However, it is unclear whether alteration of AR activity accounts for the inconsistent correlation between hirsutism and circulating androgen levels in some ethnic populations of PCOS women.
Another consideration that may contribute to androgenic effects on the hair follicle is the enzymatic conversion rate of T to its more biologically active metabolite, dihydrotestosterone. Hair growth may vary according to the activity of 5α-reductase. There are two forms (isoenzymes) of 5α-reductase, type 1 and type 2. Studies have noted that the type 2 isoenzyme predominates in hair follicles of the beard and genital hair, as well as in the testes and prostate. Unfortunately, there are no data regarding 5α-reductase activity in hair follicles of women with PCOS. Examination of 5α-reduced metabolites has shown greater production among women with PCOS compared with controls. In addition, 5α-reductase messenger ribonucleic acid (mRNA) expression in granulosa and theca cells of follicles obtained from PCOS women was higher than that observed in normal follicles. These findings are consistent with the report that genetic polymorphisms of the two isoforms of 5α-reductase were correlated with increased risk for PCOS.
Coexisting conditions that alter the bioactivity of androgens, such as hypothyroidism and obesity, may also give rise to excessive hair growth. These conditions are associated with lowered SHBG, which provides increased availability of free T. Sequence variations within the coding region of separate SHBG alleles were identified in a heterozygous woman who presented with severe hyperandrogenism during pregnancy. Serum SHBG levels were barely detectable, and non-protein-bound T concentrations were markedly higher than the normal reference range. A single nucleotide polymorphism (SNP) within an allele encoded a missense mutation that permitted normal steroid hormone binding but caused abnormal glycosylation and decreased serum SHBG.
Menstrual Irregularity
In PCOS, menstrual dysfunction is primarily characterized by irregular, infrequent, or absent menstrual bleeding. Typically, this pattern of bleeding is an extension of postmenarchal irregularity, and monthly menstrual cyclicity is never established. In some women, the onset of chronic anovulation emerges beyond adolescence, but this is unusual. Prolonged heavy bleeding should raise consideration of abnormal endometrial hyperplasia and even endometrial adenocarcinoma. About 10% of women with PCOS will exhibit regular ovulatory cycles. This is noteworthy in that a history of regular menstrual cycles does not exclude the diagnosis. It has been reported that in late reproductive life, PCOS women begin to experience regular ovulation for unknown reasons. Aging PCOS women with regular menstrual cycles appear to have a smaller follicle cohort, higher serum FSH levels, and lower androgen levels compared with age-matched PCOS with persistent anovulation. Whether changes in the follicle population or alterations in the ovarian endocrine milieu may be responsible for resumption of ovulation in late-age PCOS women remains to be determined.
Ovarian Morphology
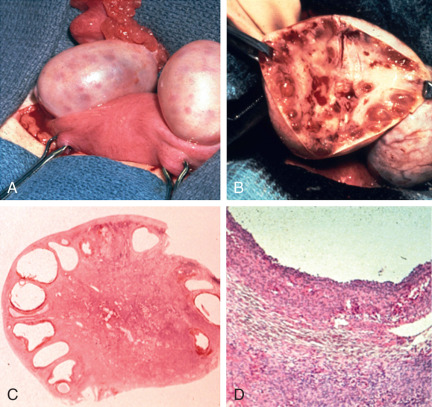
In their classical description of the polycystic ovary, Stein and Leventhal noted at surgery that the ovarian cortex contained numerous peripheral antral follicles and the ovarian stroma was enlarged comprising at least 25% of the medullary portion of the ovary. The hyperplastic medullary stroma appeared to be displacing cystic follicles towards the periphery ( Fig. 21.2 ). Subsequent introduction of ultrasound imaging facilitated the clinical identification of polycystic ovaries, although morphological criteria were not uniform.
In 2003, description of the polycystic ovary was established ( Fig. 21.3 ) at a consensus conference in Rotterdam, The Netherlands. A polycystic ovary was defined as having 12 or more follicles per ovary, irrespective of location, or an ovarian volume of greater than 10 cm 3 . Currently this definition has been uniformly accepted. However, with the advent of newer equipment and advanced technology, the precision of counting follicles has improved. A recent task force report recommended that the threshold for polycystic ovary morphology based on follicle count should be increased to greater than 25 follicles per ovary to avoid overinterpretation of polycystic ovary morphology and misdiagnosis of PCOS. This new recommendation warrants standardization of follicle counting techniques and validation of the threshold defining follicle excess. Moreover, the utility of this new recommendation in clinical practice should be assessed. Until these measures are defined, the current ultrasonographic criteria or 12 or more follicles per ovary or a volume greater than 10 cm 3 is suitable.
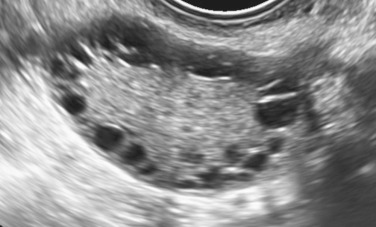
This description of the polycystic ovary should not be confused with the ultrasound appearance of an ovary in women recovering from hypogonadotropic hypogonadism. In these individuals multiple small antral follicle growth is common and may appear similar to a polycystic ovary. In women undergoing ovulation induction, multiple follicle formation may occur as result of ovarian stimulation. Thus implication of a multifollicular ovary or polycystic ovary should be considered within the realm of the clinical setting.
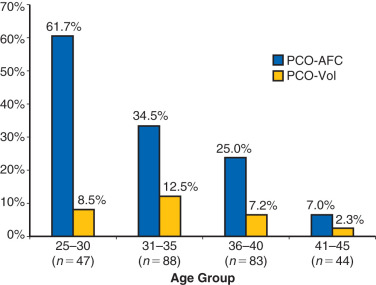
While the sonographic demonstration of polycystic ovaries in the presence of typical clinical features is regarded as confirmatory for the syndrome, it has been clearly demonstrated that polycystic ovary morphology can be found in normal ovulatory women without a history of hyperandrogenism. In a study of 104 young Danish women with polycystic ovaries, only 43 were found to have other symptoms that fulfilled the criteria for PCOS. Thus 58% of women with polycystic ovaries were essentially normal without menstrual abnormalities or evidence of hyperandrogenism. Similarly, polycystic ovaries were recently shown to be a relatively common finding in a large series of well-characterized normal women from age 25 to 45 years old. Of 257 women, polycystic ovaries were identified in 32% overall with the greatest percentage, 62%, occurring in women 25 to 30 years old followed by progressive decline in older groups ( Fig. 21.4 ). The physiological relevance of polycystic ovaries in normal women is not clear, although a longitudinal study suggested that such individuals are not at increased risk for PCOS. Thus, while the requisite identification of polycystic ovaries to establish the diagnosis appears to be clinically appropriate, the morphogenesis of the polycystic ovary may not necessarily be unique to the disorder.
Obesity
The prevalence of obesity as a worldwide epidemic has dramatically increased over the past two decades. In the United States, almost two-thirds of women are overweight or obese, as are about one-half of Australian women. Similar increasing proportions of overweight and obese people have occurred in other countries, with the prevalence of obesity increasing also in reproductive-aged individuals. Obesity is a disease of excess body fat that is closely related to insulin resistance. Obesity increases the risks of hypertension, dyslipidemia, diabetes, and cardiovascular disease and elevates the rate of all-cause mortality.
Although the prevalence of obesity in women with PCOS also has increased over time, obesity per se is not an intrinsic defect to PCOS, particularly since 40% to 50% of PCOS women are not obese. Rather, obesity interacts with hyperandrogenism to worsen the PCOS phenotype or perhaps promote the development of PCOS in susceptible individuals. It is unclear whether PCOS women are predisposed to obesity. Postprandial thermogenesis may be decreased in PCOS, but resting energy expenditure in PCOS appears equivalent to that of normal weight-matched control. Interestingly, nonobese PCOS women show diminished use of lipid as a metabolic substrate during overnight fasting compared with normal women, suggesting that impaired conversion of lipid to carbohydrate/protein metabolism in PCOS women may underlie enhanced weight gain ( Fig. 21.5 ).
Women with NIH-defined PCOS preferentially accumulate abdominal fat with weight gain, giving rise to a greater waist to hip ratio than BMI-matched normal women. This type of increased abdominal fat distribution, also termed android obesity, promotes insulin resistance through increased intraabdominal (visceral) fat, occurs in overweight/obese with PCOS and other hyperandrogenic states, and accompanies metabolic syndrome as a risk factor for cardiovascular disease (see later). Whether PCOS women with less degrees of adiposity have similar, but more subtle, changes in body fat distribution is unclear, since a study of PCOS women and control women matched for BMI and fat mass showed similar regional fat distribution (i.e., subcutaneous [SC] abdominal, intraabdominal and gluteofemoral fat stores) between female groups despite insulin resistance in PCOS women. More recently, however, sophisticated radiographic imaging techniques in PCOS women have shown increased abdominal body fat underlying hyperinsulinemia from insulin resistance over a wide BMI range, accompanied by hyperandrogenism, consistent with greater intraabdominal fat mass in normal-weight PCOS women than age- and BMI-matched control women ( Table 21.2 ). In contrast, normal women with gynecoid obesity generally have an enhanced accumulation of fat in the hip, buttock, and thigh regions.
| Total Body DXA | NL ( n = 13) | PCOS ( n = 6) | P Value |
|---|---|---|---|
| Total body fat (kg) | 19.8 ± 0.9 | 21.2 ± 1.7 | 0.5 |
| Total body fat (%) | 31.5 ± 0.8 | 34.0 ± 1.3 | 0.1 |
| Total body lean mass (kg) | 40.4 ± 1.3 | 39.8 ± 2.2 | 0.8 |
| Android fat (kg) | 1.1 ± 0.1 | 1.5 ± 0.2 | 0.02 |
| Android fat (%) | 5.5 ± 0.2 | 7.1 ± 0.5 | 0.02 |
| Gynoid fat (kg) | 4.3 ± 0.2 | 4.4 ± 0.3 | 0.9 |
| Gynoid fat (%) | 21.9 ± 0.4 | 20.8 ± 0.4 | 0.08 |
| Abdominal MRI | NL ( n = 14) | PCOS ( n = 6) | P Value |
| SC abdominal fat (kg) | 4.4 ± 0.2 | 5.0 ± 0.4 | 0.2 |
| Intraabdominal fat (kg) | 1.8 ± 0.1 | 2.4 ± 0.3 | 0.03 |
Insulin Resistance
It has been well-documented that women with PCOS are insulin resistant and have compensatory hyperinsulinemia as a result of their disorder. The prevalence of glucose intolerance or type 2 diabetes in PCOS has been reported to be about 40%, depending upon the population studied. That insulin resistance is common in obesity may account, in part, for the rather wide prevalence. Nevertheless, independent of obesity, the presence of a defect in insulin action in PCOS has been clearly established. Generally, the degree of insulin resistance is mild, although the prevalence of glucose intolerance and subsequent diabetes has been reported to be as high as 31% and 7.5%, respectively. Moreover, in these individuals, the progression to diabetes appears to be relatively rapid. Commonly, fasting hyperglycemia is not evident; instead, there is a primary dysfunction of postprandial glucose uptake that results in severe peripheral insulin resistance.
Not only are women with PCOS predisposed to insulin resistance and risk for diabetes, but also evidence exists that first-degree relatives exhibit disordered glucose metabolism and insulin secretion. Importantly, this predisposition includes both sisters and brothers of affected individuals. Aside from the increased risk for diabetes, there is indirect evidence to indicate that insulin resistance may worsen the clinical manifestations of PCOS. Administration of insulin-lowering drugs has been shown to improve insulin sensitivity, reduce androgen levels, and restore ovulation in some, but not all patients with this disorder. Insulin resistance may also contribute to metabolic dysfunction in PCOS, including an increased likelihood of lipid abnormalities. In addition, the association of insulin resistance with visceral fat distribution is underscored by the displacement of central fat to the peripheral compartment with improvement of insulin sensitivity following administration of insulin-lowering drugs or weight reduction.
In addition to the increased prevalence of impaired glucose tolerance and risk of type 2 diabetes in women with PCOS, the rate at which the disease progresses appears to be accelerated. Several studies have demonstrated that conversion of impaired glucose tolerance to type 2 diabetes is enhanced in these individuals. This process is increased in the presence of obesity or a family history of type 2 diabetes.
Depression and Anxiety Disorders
Recent studies have begun to explore the relationship of PCOS to quality-of-life issues including mood and anxiety disorders. Most studies comprise meta-analyses that reflect an increased frequency of depression and anxiety. In a meta-analysis of 12 studies, women with PCOS had higher depression and anxiety scores compared with controls. Consistent with these findings, a large meta-analysis of 28 studies found a higher prevalence of depression and anxiety among women with PCOS than that of controls. In addition, quality-of-life issues were also lower in the PCOS women. However, in these studies, it was emphasized that the degree of emotional distress was mild and did not necessarily exceed the normal range. In contrast, in a systematic review by Dokras et al., women with PCOS had substantially increased mood and anxiety disorders compared with age-matched controls. These findings were consistent with the observation that women with PCOS displayed higher rates of depression, anxiety, and social phobia than controls. In a recent nationwide, population-based cohort study comprising 5431 women with PCOS and 21,724 matched controls without PCOS, the risk of newly diagnosed depression and anxiety disorders after entry into study was slightly higher in the group of PCOS women. Collectively, these results appear to suggest that women with PCOS are at increased risk for mood and anxiety disorders, although their relationships to PCOS and/or its comorbidities warrant further investigation.
Acne and Acanthosis Nigricans
Women with PCOS may experience increased skin oiliness secondary to excessive stimulation of the pilosebaceous unit by increased androgen production. However, increased sebaceous gland activity in PCOS is not associated with acne nor is acne correlated with increased ovarian androgen production. Therefore, as an isolated symptom, acne should not be considered a sign of PCOS.
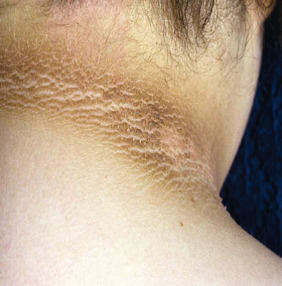
Perhaps more common is the finding of acanthosis nigricans, which has been observed in 5% to 50% of hyperandrogenic women and is related to the presence and severity of hyperinsulinemia. This dermatological condition features symmetrical, darkened, velvety plaques that may appear most commonly on the nape of the neck, in the intertriginous areas of the body such as skin folds, and on pressure-bearing surfaces such as knuckles and elbows ( Fig. 21.6 ). Acanthosis nigricans originates from epidermal hyperkeratosis and dermal fibroblast proliferation. There is no evidence of an increased number of melanocytes or melanin deposition despite apparent increased pigmentation. Acanthosis nigricans is considered a potential marker for insulin resistance and diabetes in adults. Reduction of hyperinsulinemia is associated with improvement in the darkened skin areas.
Infertility
PCOS is the leading cause of anovulatory infertility. Approximately 75% to 85% of PCOS women have some degree of ovulatory dysfunction, although ovulation may occasionally occur in these individuals. Equally important, PCOS women with regular menstrual cycles also can be anovulatory, as shown in a study by Chang et al., in which 16% of 316 PCOS women with hyperandrogenism and oligo-anovulation had normal-appearing menstrual cycles (i.e., 27- to 34-day intervals).
PCOS also is often accompanied by obesity as an additional risk factor for anovulation. Although the endocrine events underlying ovulatory dysfunction in obese PCOS women remain unclear, PCOS and obesity likely interact to disrupt ovulation, with normal-weight and obese PCOS patients representing different ends in a continuum of ovulatory dysfunction. In normal-weight PCOS patients, LH hypersecretion and ovarian hyperandrogenism appear to be the primary factors governing altered follicle development, with insulin also enhancing gonadotropin actions on ovarian steroidogenesis and granulosa cell differentiation. In obese PCOS patients, on the other hand, metabolic factors, including abnormal glucose-insulin homeostasis and adipogenic dysfunction, may be more important than LH in altering follicle development and oocyte maturation, explaining why weight loss can restore ovulation and fertility in many obese PCOS patients but not in normal-weight PCOS patients in whom ovulation induction is required.
Fetal Origin of Polycystic Ovary Syndrome
- ◆
Alterations of the maternal-fetal environment may predispose to PCOS after birth.
- ◆
Fetal exposure to androgen or insulin excess may provoke an adult PCOS phenotype.
Alterations in the maternal-fetal environment can permanently program adult disease through epigenetic modifications of genes that influence fetal susceptibility to disease after birth. For example, women with congenital adrenal virilizing tumors or classical CAH from 21-hydroxylase deficiency have an increased risk of developing a PCOS-like syndrome in adulthood, implicating female fetal androgen excess as a modifier of PCOS phenotypic expression after birth. This relationship between hyperandrogenism in utero and PCOS phenotypic expression in adulthood, however, remains controversial since women born from opposite sex twins do not show an increased prevalence of PCOS-like features, assuming they share a prenatal environment with a male cotwin that increases their exposure to androgen. Furthermore, the length of the second finger relative to the fourth finger, as an anatomical marker of in utero androgen exposure, is altered in some, but not all, PCOS women, as it is in adult prenatally androgenized female rhesus monkeys with PCOS-like features. Elevated umbilical cord T levels also occur in some, but not all, newborns of PCOS mothers, perhaps due to variability in placental steroidogenesis or differences in cord blood collection at term, a time point beyond the critical period of human ovarian differentiation.
Perhaps more importantly, amniotic fluid T levels in the second trimester are normally higher in the male than the female fetus, allowing a wider range of T levels for fetal sexual differentiation than term umbilical vein T levels that are similar between sexes. This is because a transient rise of pituitary gonadotropins at midgestation increases androgen production by the testes compared with the ovary, temporarily elevating circulating androgen levels in the male compared with the female fetus. In this regard, 40% of midgestational female fetuses have elevated serum androgen levels into the normal male range. Furthermore, amniotic fluid T levels are elevated in female fetuses of PCOS mothers compared with those of normal mothers, suggesting that hyperandrogenism could influence developmental programming of the female fetus, assuming a critical second trimester time interval when a susceptible fetus is exposed to androgen excess ( Table 21.3 ).
| N | Mean | SD | |
|---|---|---|---|
| Control mothers | |||
| Male fetus | 21 | 0.80 * | 0.16 |
| Female fetus | 24 | 0.36 | 0.10 |
| PCOS mothers | |||
| Male fetus | 17 | 0.87 * | 0.21 |
| Female fetus | 13 | 0.53 † | 0.12 |
* P < .001 fetal sex difference.
In support of this, the midgestational human fetal ovary has several steroidogenic enzymes; genes encoding multiple steroid signaling pathways; and receptors to steroids, insulin, insulin-like growth factor (IGF)-I and IGF-II. It also has the capacity to produce androgens due to the presence in stroma and theca-like cells containing 17a hydroxylase-17, 20-lyase (P450c17), the major enzyme responsible for androgen production. As evidence, cultured human fetal ovaries at this gestational age can metabolize pregnenolone sulfate to DHEA and androstenedione (A) and can secrete in decreasing amounts DHEA, progesterone and estrone; with less amounts of A, estradiol, and T. Although lacking functional LH-like receptors, the midgestational human fetal ovary may produce androgens in vivo in response to insulin, since previous studies have shown elevated amniotic fluid T levels in diabetic women along with hirsutism, ovarian theca-lutein cysts, and thecal cell hyperplasia in their female stillbirth offspring.
But what is the link between the maternal environment and androgen excess in utero? Although maternal serum androgen levels in midgestation are greater in PCOS than normal women, maternal androgens are unlikely to directly program PCOS in offspring if placental aromatization is normal. Alternatively, an increased risk of maternal glucose intolerance in PCOS women may induce hyperglycemia in utero, which may stimulate fetal insulin release to promote androgen production and/or folliculogenesis in the fetal ovary. As an example, prenatal T administration to female rhesus monkeys impairs maternal-fetal glucose-insulin homeostasis and stimulates fetal insulin release, consistent with other animal models linking maternal metabolic dysfunction and fetal androgen excess with adult PCOS-like phenotypes.
Such maternal-fetal environment dysfunction may underlie several endocrine antecedents of PCOS. Infant girls born to PCOS mothers exhibit anti-müllerian hormone (AMH) overproduction as a marker of growing follicles, which persists into prepubertal life and improves when PCOS mothers receive metformin in pregnancy, beginning at or before conception. Also, serum leptin levels in newborns of PCOS women positively correlate with birth weight and maternal BMI at midgestation, while enlarged ovaries and hyperinsulinemia in female children of PCOS women coexist at puberty with LH hypersecretion and androgen excess. Low infant birth weight associated with PCOS pregnancies in Chilean and Iranian women and precocious puberty accompanying PCOS in northern Spanish women implicate impaired fetal nutrient availability from placental insufficiency. However, developmental programming effects on the fetus after birth can occur despite normal infant birth weight. For example, prenatal androgenization of female rhesus monkeys impairs fetal glucose-insulin homeostasis without affecting infant birth weight and exaggerates neonatal growth as antecedents to an adult PCOS-like phenotype consisting of disordered SC abdominal and visceral adipogenesis.
Polycystic Ovary Syndrome in Adolescence
- ◆
Hyperandrogenism is likely the best indicator of PCOS in adolescence.
- ◆
Persistent irregular bleeding beyond 2 years after menarche is consistent with PCOS.
- ◆
Obesity in adolescence increases severity of the syndrome.
- ◆
Insulin resistance and fatty liver are common.
- ◆
Premature pubarche increases the prevalence in adolescence.
Diagnostic Criteria
Diagnostic criteria for PCOS in adolescence have not been thoroughly established. While adolescent girls exhibit similar clinical features of the disorder as those by adult women, interpretation of significant symptomatology warrants further study.
Evidence of Androgen Excess
Perhaps the most reliable indicator of PCOS in adolescence is clinical hyperandrogenism, as evidenced by hirsutism. While quantification of excess hair growth in adult women has been determined by F-G scoring, there is little information about hirsutism in adolescent girls. In a study of 856 black and white girls, ages 9 and 10, that were followed for 9 years, the presence of upper lip hair was assessed by F-G scoring. In postmenarchal girls the percent of those with grade 1 hair growth was 15.8%, grade 2, 12.4%, grade 3, 2.1%, and grade 4, 0.6%. Notably, this prevalence is similar to that reported in 430 consecutive women between the ages of 15 to 24 attending a general medical outpatient clinic. This suggests that F-G scoring may be used to quantify adolescent hair growth. Interestingly, in an analysis of hair growth in hirsute and nonhirsute adult Norwegian and Dutch women, it was demonstrated that the sum of scores from the lip, chin, and pubic regions were sufficient to discriminate between those with and without hirsutism. These body areas are easily accessible and may provide for convenient clinical assessment of hair growth in adolescent girls in an office setting.
It is important to emphasize that in adolescence isolated mild hair growth should not be considered as clinical evidence of hyperandrogenism. However, mild hirsutism accompanied by abnormalities of reproductive or metabolic function may warrant further evaluation.
Evidence of androgen excess also includes elevated serum total or free T levels. In early adolescence circulating T gradually increases to achieve adult levels by 2 to 3 years following menarche. This pattern was observed in girls with regular and irregular menstrual cycles as well as those with oligomenorrhea. In the presence of obesity, both total and free T levels were markedly increased during puberty and early adolescence. Enhanced T secretion was associated with hyperinsulinemia that likely amplified theca cell androgen production and lowered SHBG. Unfortunately, few studies have carefully examined variable T secretion in puberty and early adolescence. Recently, serum T levels, assessed by liquid chromatography-tandem mass spectrometry, were reported for late-age adolescent women (median age of 17.5 years). Notably, the upper limit of values for T was comparable to those observed in normal adult women. These findings suggest that normative T values for adult women may be used to determine hyperandrogenemia in late-age adolescence, although in early adolescent girls with suspected PCOS discriminatory T values may be lower.
Acne has been associated with hyperandrogenism and may reflect increased androgen production in adolescent PCOS. However, acne is common in adolescent girls with a reported frequency of greater than 50%. The basis for acne in normal puberty has been attributed to adrenal androgen production. Because acne is commonly observed in puberty and early adolescence, it is not included in the criteria for PCOS. Nevertheless, in cases of moderate to severe acne, assessment of circulating androgen levels is reasonable.
Irregular Menstrual Bleeding
Unlike adult women, adolescent girls frequently experience menstrual irregularity following menarche and during early adolescence. Several attempts to document menstrual bleeding and ovulation rates in this population have revealed highly variable patterns among individuals. During the first 2 years postmenarche, anovulation is common and marked by irregular bleeding. By the third year, regular cyclicity was observed in 95% of adolescents with an average of 10 or more cycles per year. Conversely, approximately 50% of 14- to 16-year-old girls with oligomenorrhea had persistence of irregular cycles at age 18. Similar findings were exhibited by young adolescent girls with oligomenorrhea followed for up to 6 and 8 years. Collectively, these data suggest that in adolescence, at least 2 years beyond menarche, a pattern of irregular menstrual bleeding accompanied by evidence of androgen excess is consistent with a diagnosis of PCOS.
Ovarian Morphology
Unlike imaging criteria for polycystic ovarian morphology for adult women, characteristic features of a polycystic ovary have not been established in adolescent girls with PCOS. This is due, in large part, to developmental changes of the ovary during puberty. Notably, it has been well demonstrated in normal, healthy girls that the ovary may exhibit more than 12 follicles per ovary as determined by ultrasonography. These data suggest that polycystic ovaries are relatively common in the normal adolescent population. Thus the antral follicle count appears to be an unreliable measure suggestive of PCOS in adolescence. Instead, most studies have shown that ovarian volume has a steady growth pattern throughout puberty and adolescence. As a result, it has been recommended that ovarian volume be used to assess ovarian morphology. Although most studies have relied on a value of greater than 10 cm 3 to detect polycystic ovary morphology, a recent report by a group of experts convened by the Pediatric Endocrine Society recommended a value of greater 12 cm 3 as sufficient for ovarian enlargement.
In an effort to achieve a more precise image of the ovary in adolescent girls with possible PCOS, investigators have examined the use of magnetic resonance. Brown and coworkers described ovarian morphology in normal high school students and adolescents with PCOS based on the presence of hirsutism and oligomenorrhea. Employing magnetic resonance imaging, 17 of 21 normal individuals (81%) had 12 or more follicles per ovary ( Fig. 21.7 ). In 5 of 21 normal adolescents ovarian volume was larger than 10 mL. Similar findings were reported by Kenigsberg et al. for 39 PCOS and 22 normal girls matched for age and BMI who underwent MR imaging. Most of the subjects were obese. In the PCOS group, the median ovarian volume and follicle number per ovary were 11.8 cm 3 and 12, respectively, both of which were significantly greater than those of normal controls. Moreover, in a comparison of techniques, ovarian volume by MR imaging was significantly larger than that assessed by ultrasound, whereas comparison of follicle number was not possible due to a lack of clear imaging by ultrasound. It is apparent that MR imaging provides clear depiction of the ovary unencumbered by obesity; however, cost and inconvenience of this technique together with a lack of normative data in adolescents limits recommendation.
It has been proposed that serum AMH may be used as a surrogate measure for antral follicle number in adults. In healthy girls with polycystic ovary morphology, AMH levels are elevated. However, the utility of AMH among adolescents with PCOS has not been established.
Clinical Presentation
Characteristically, during puberty or early adolescence, the symptoms of PCOS may emerge insidiously coincident with changes that accompany normal pubertal development (see Chapter 18 ). That the emergence of PCOS can commonly be traced to the events of puberty suggests that this disorder may be related to an abnormal expression of those factors that initiate and regulate the process of puberty. Because the duration of menstrual irregularity that accompanies normal puberty may be variable, it is difficult to rely solely on this historical feature as a basis for diagnosis. Moreover, with the recognition that some PCOS women may exhibit normal ovulatory function, evidence of regular cyclic bleeding does not preclude the disorder in adolescence. Rather, early detection of PCOS in adolescent girls is predicated primarily on hyperandrogenic symptoms such as hirsutism and acne. In the obese individual, associated metabolic-reproductive abnormalities may create uncertainty as to the mechanism of hyperandrogenism. Reduction in SHBG is directly correlated to obesity, thus giving rise to increased free testosterone levels. In addition, obese adolescents, particularly those with evidence of acanthosis nigricans, are highly likely to have insulin resistance with compensatory hyperinsulinemia, which is known to suppress SHBG and contribute to excess ovarian androgen production. An ultrasound image of a polycystic ovary as determined by size or follicle number may be helpful with the diagnosis. However, the utility of ultrasonography in adolescent girls is limited by the lack of established morphological criteria.
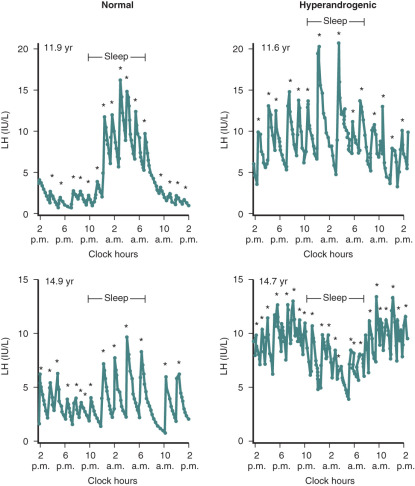
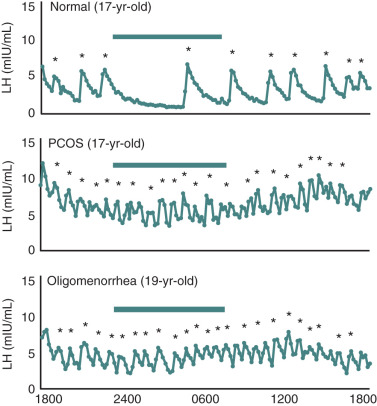
It has been well-documented that adolescent girls with PCOS have increased levels of circulating androgens as well as elevated LH levels. Previous studies have shown that hyperandrogenic girls with likely PCOS exhibited changes in gonadotropin secretion patterns that were similar to those found in adult PCOS. Increased concentrations of serum LH are accompanied by an increase in pulse frequency and amplitude. In addition, mean serum levels of testosterone and androstenedione were elevated. Twenty-four-hour pulsatile LH secretion studies have revealed that premenarchal hyperandrogenic girls demonstrated higher LH levels while awake whereas the sleep entrained increases were minimal compared with those of developmentally matched control subjects. In postmenarche, there was greater LH pulse activity during waking hours whereas pulse frequency was slowed with sleep ( Fig. 21.8 ). By comparison, in postmenarchal normal girls, LH levels were lower and the pulse frequency reduced while awake whereas during sleep LH pulse frequency was significantly less than that of hyperandrogenic girls. Thus the pattern of gonadotropin secretion in the postmenarchal normal control resembled that of the younger premenarchal hyperandrogenic girl. These findings suggest that the transition through neuroendocrine puberty may be accelerated in hyperandrogenic girls compared with normal puberty. Moreover, the data pose the question as to whether there is a chronological difference in the onset of increased LH levels and pulse frequency between hyperandrogenic and normal pubertal girls.
Among postmenarchal girls with irregular bleeding, it has been estimated that approximately 50% of oligomenorrheic adolescent girls have increased levels of serum LH associated with mild elevations of circulating androgens. In addition, we and others have shown that these individuals exhibited an increased rate of LH release as determined from frequent sampling studies, which suggested a diagnosis of PCOS ( Fig. 21.9 ). Notably, in a long-term follow-up study of oligomenorrheic girls, those with normal serum LH values eventually developed regular ovulatory function compared with more than half of those with elevated LH levels in which the gonadotropin abnormalities persisted along with hyperandrogenism. These intriguing findings suggest that oligomenorrhea in early adolescence may be associated with an endocrinologic phenotype of PCOS in the absence of overt signs of hyperandrogenism. Whether the transitory nature of elevated LH and androgens represents an extension of normal hypothalamic-pituitary development remains to be determined.
Role of Obesity
The effect of pubertal obesity on the human hypothalamo-pituitary-ovarian axis has been examined extensively in nonobese and obese girls. Throughout puberty (i.e., Tanner stage 1–5), obese girls exhibit hyperinsulinemia and hyperandrogenism (i.e., increased total T, decreased SHBG, and elevated free T levels). Simultaneously, they also show reduced LH pulse amplitude compared with that of nonobese girls that resembles the inverse relationship between BMI and serum LH levels in adult PCOS women, likely due to reduced pituitary responsiveness to gonadotropin releasing hormone (GnRH) and/or increased LH clearance. Similarly, obese girls exhibited lower LH pulse frequency compared with nonobese girls in early puberty. With advanced puberty, pulsatile LH release increased in obese girls and was comparable or slightly greater than that of nonobese girls.
During early-stage puberty, the diurnal, sleep-entrained pattern of LH secretion is dampened by obesity. With further pubertal development (Tanner 3–5), obese adolescents exhibit an increase in 24-hour LH pulsatile secretion that is greater than that of nonobese individuals and coincident with an exaggerated rise in circulating free T levels. This finding resembles the heightened pattern of LH secretion seen in PCOS women. These findings imply that as hypothalamic GnRH activity increases throughout puberty, obesity-related hyperandrogenism reduces hypothalamic steroid negative feedback to progesterone and promotes LH hypersecretion.
In support of this concept, progesterone administration to adolescent hyperandrogenic girls often fails to reduce LH release. Similarly, progesterone administration to PCOS women is unable to normally suppress LH pulse frequency but can do so with coadministration of the antiandrogen, flutamide, which restores hypothalamic steroid negative feedback to progesterone.
Abnormal Metabolic Function
Similar to adults, adolescent girls with hyperandrogenic PCOS exhibit abnormal insulin responses to glucose loading. Correspondingly, assessment of 24-hour patterns of insulin revealed greater release in hyperandrogenic girls than that observed for normal girls, whereas a reciprocal relationship was found for IGFBP-1 secretion. The presence of obesity only worsens the likelihood of insulin resistance. It has been reported that of the endocrine obesity syndromes in adolescence, PCOS is the most common. While obesity contributes to insulin resistance, it has been shown that obese, hyperandrogenemic PCOS girls have higher fasting serum insulin, less insulin sensitivity, and greater insulin secretion compared with obese girls without androgen excess. These findings are supported by studies in adolescent PCOS, which demonstrated that increased free T levels predicted the presence of insulin resistance independent of BMI. The lipid profile in PCOS girls with increased androgen indices have demonstrated higher low-density lipoprotein cholesterol (LDLC) to high-density lipoprotein cholesterol (HDLC) ratios in conjunction with lowered SHBG levels. This adverse lipid profile is underscored by obesity. However, associated comorbidities may vary among obese individuals as some have considerably lower risk. Recently, it has been shown that metabolically healthy, obese PCOS girls exhibited favorable risk for type 2 diabetes, atherogenesis, inflammation, and hyperandrogenemia compared with obese adolescent PCOS with unhealthy metabolic profiles.
Obesity also confers risk for fatty liver disease, particularly in those with insulin resistance. Studies in adult women with PCOS have shown that insulin resistance is the primary contributor to nonalcoholic steatohepatitis. In obese adolescents with PCOS, the prevalence of nonalcoholic steatohepatitis, based on serum transaminase levels, was reported to be 43.6%. These data suggest that long-term health consequences associated with abnormal metabolic function may be established early in reproductive life for girls with PCOS.
Prepubertal Disposition
It has been reported that girls with premature pubarche are at increased risk for functional ovarian hyperandrogenism and polycystic ovaries following puberty (see Chapter 17 ). Moreover, with subsequent development of hyperandrogenism and hyperinsulinemia, there was a corresponding reduction in birth weight of these individuals. The link between low birth weight and insulin resistance in children appears to be persistent throughout life as indicated by studies performed in early and late adulthood. This relationship may be of particular relevance to a possible mechanism for PCOS in this population. Low birth weight is commonly associated with hypoplasia of the fetal adrenal and correspondingly low serum DHEA-S levels. DHEA-S secretion also serves as a marker for adrenarche, which is independent of and precedes gonadarche by several years. It has been demonstrated in pairs of siblings that were discordant at birth but of equivalent weight during childhood that DHEA-S levels were higher in those of low birth weight compared with those born with normal weight. Thus, if as proposed, fetal growth modulates adrenarche, then increased DHEA-S may have reflected an exaggerated adrenarche in these children. The resultant increased androgen pool may set in motion a cycle of altered physiology characteristic of PCOS. This notion is further reinforced by the presence of hyperinsulinemia and insulin resistance, which may enhance androgen production in adolescent girls at risk for PCOS. In addition, elevated levels of circulating insulin coincide with a reciprocal decline of SHBG, thereby allowing for increased availability of free T. Thus the detection of hyperinsulinemia in postmenarchal girls with hyperandrogenism relates temporally to physiological insulin resistance during puberty and may be of critical importance in the genesis of PCOS.
While premature pubarche may increase the risk for PCOS after puberty, exposure to androgen prepubertally may also provoke features of the disorder. In a study conducted in nonhuman primates, prepubertal female rhesus macaques at 1 year of age were administered Silastic T implants resulting in fourfold rise of circulating levels. This level of induced hyperandrogenemia was maintained long-term throughout the course of study. At age 5 when the monkeys were in early adulthood, T-treated animals had a significantly greater LH pulse frequency during the early follicular phase compared with controls given cholesterol-filled implants. No differences were seen in insulin sensitivity, although there was suggestion of multiple follicle formation in the ovaries as determined by magnetic resonance. Ovulation rates were similar in both groups. These results show that increased androgen exposure over the course of pubertal development may enhance the neural drive to the reproductive axis that resembles those of obese hyperandrogenemic girls in early adulthood and are characteristic of PCOS. Moreover, the findings suggest that the prepubertal hypothalamic-pituitary-ovarian axis may be vulnerable to endocrinologic insult such as excess androgen production.
Altered Physiology
- ◆
Ovarian follicle growth is arrested at the mid-antral stage of development in PCOS.
- ◆
Hyperandrogenemia reduces pituitary responsiveness to progesterone negative feedback leading to increased LH secretion.
- ◆
Androgen excess is primarily derived from altered theca cell steroidogenesis.
- ◆
Despite follicular arrest, granulosa cells exhibit enhanced responsiveness to FSH that increases the risk of ovarian hyperstimulation.
- ◆
Metabolic dysfunction includes abnormal SC adipocyte function, defective pancreatic beta cell function, and impaired insulin signaling.
A clear explanation of the pathogenesis for PCOS has not been established. The clinical presentation may be variable, as diagnostic subgroups have been defined. Nevertheless, the predominant clinical features of androgen excess and anovulation are commonly accompanied by inappropriate gonadotropin secretion, abnormal steroid production, increased insulin secretion, dyslipidemia, and obesity. Mechanistic integration of these factors has been proposed, yet these concepts await confirmation.
Oocyte Competence
Within the changing microenvironment of the developing follicle, bidirectional cumulus cell-oocyte signaling governs the gradual acquisition of developmental competence by the oocyte, defined as the ability of the oocyte to complete meiosis and undergo fertilization, embryogenesis, and term development ( Fig. 21.10 ) (see Chapter 8 ). Studies of oocyte developmental competence in PCOS women, however, are limited for several reasons, including ethical and experimental constraints on using human oocytes and embryos for biomedical research. The unique microenvironment of each follicle also has its own effect on the developing oocyte so that transferring multiple embryos into the uterus makes it difficult to monitor relationships of follicle fluid substances with embryo implantation. Another problem is the gradual acquisition of necessary molecular components to complete meiosis, preimplantation embryogenesis, and successful embryo implanation, limiting understanding of oocyte development in PCOS to indirect markers, including studies of oocyte gene expression, correlations of follicle fluid hormone and metabolite levels with oocyte development in vivo, and actions of sex steroids on oocyte development in vitro.
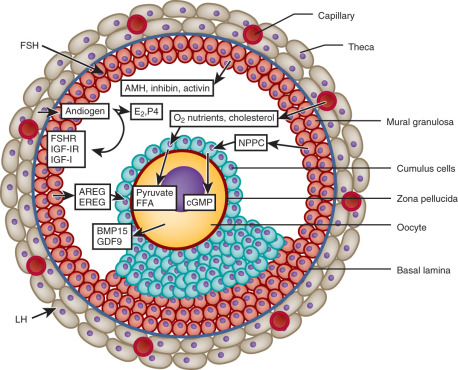
Given these limitations, oocyte development is influenced in part by phenotypic expression of PCOS by Rotterdam criteria. Women with PCOS by NIH criteria, for example, have the greatest degrees of hyperandrogenism and hyperinsulinemia compared with women with either ovulatory or nonhyperandrogenic PCOS and are the most likely individuals to have an abnormal intrafollicular environment leading to impaired oocyte developmental competence. During ovarian stimulation for in vitro fertilization (IVF), hyperandrogenic follicles of PCOS women by NIH criteria contain normal-appearing metaphase II oocytes with abnormal gene expression that affects signal transduction, cell metabolism, DNA transcription, and RNA processing. These differentially expressed oocyte genes often contain promoter sequences with putative binding sites for AR and PPAR-γ, linking androgen excess with insulin resistance at the level of the oocyte. Consequently, obese PCOS women (84% of whom fulfill NIH criteria) have a lower chance of successful pregnancy outcome by IVF than lean PCOS women (65% of whom fulfill NIH criteria). Conversely, nonobese IVF patients with PCOS by Rotterdam criteria have clinical pregnancy and live birth rates/cycle that are similar to those of nonobese IVF patients with male factor infertility, although their oocytes are smaller in size (112 µm) and less likely to mature (71%) than oocytes of the latter individuals (oocyte size, 117 µm; percent oocyte maturity, 83%).
Impaired oocyte developmental competence, however, is related to several factors. Impaired oocyte competence has been linked with elevated follicle fluid levels of tumor necrosis factor alpha and interleukins. It also may involve excess cellular lipid accumulation within the cumulus cell-oocyte complex, as shown in mice fed a high fat diet, whereby poor oocyte quality and abnormal embryogenesis are associated with increased lipid content and oxidative stress, reduced mitochondrial function, and increased apoptosis within these cells. The degree to which inflammation plays a role in this process remains to be determined.
Evidence that women with PCOS have a higher incidence of spontaneous pregnancy loss is inconsistent. In one report, the prevalence of polycystic ovaries in women with recurrent miscarriage was 56%, while in another, polycystic ovarian morphology was not predictive of pregnancy loss among such women. Although still debated as to whether PCOS per se is directly linked with miscarriage, PCOS does not appear to be an independent risk factor for miscarriage, which instead is strongly associated with BMI.
Disordered Folliculogenesis and Ovarian Morphology
The process that leads to the distinctive morphological features of the polycystic ovary is currently not understood. It appears as if normal follicular growth occurs up to the midantral stage, after which maturation ceases and the majority of follicles progressively undergo atresia. At the midantral stage of follicle development, the granulosa cell layer becomes progressively degenerative, and the entire structure may have the appearance of a thin-walled cyst ( Fig. 21.11 ). However, that follicle development becomes arrested at the midantral stage does not necessarily signal the immediate onset of atresia. In granulosa cells from unstimulated ovaries of women with PCOS, viability measures have indicated robust cell survival with substantial steroidogenic potential compared with cells obtained from ovaries of normal women. It was concluded that the majority of follicles in PCOS are not apoptotic but retain ample functional capacity. These findings were consistent with a study in which PCOS granulosa cells exhibited decreased apoptotic nuclei and proteins and increased antiapoptotic protease gene expression compared with that of normal cells. After midantral arrest, there appears to be a progressive accumulation of follicular fluid that results in expansion of the antrum, which gives rise to the classical appearance of cystic follicles. As the follicle enlarges, the granulosa cell layer becomes progressively degenerative and thin.
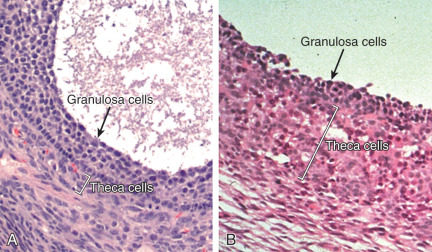
In contrast to granulosa cells, the theca cell layer that surrounds the follicle becomes substantially hyperplastic and thickened compared with that of normal follicles and is responsible for increased androgen production (see Fig. 21.11 ). The mechanism responsible for the unusual proliferation of theca cells in this disorder is unknown.
Follicle number is increased in PCOS ovaries at the primary, secondary, and early antral stages of growth compared with that observed in normal ovaries. The basis for numerical disparity in follicle populations is not clear. Webber et al. demonstrated that in polycystic ovaries, the ratio of primary to primordial follicles was increased, which may have reflected enhanced entry into the growth pool by primordial follicles. In contrast, Maciel showed that increased number of growing follicles was not associated with a reduction of primordial follicles and suggested that the accumulation of growing follicles may result from restricted growth progression.
These observations raise the question of whether in PCOS extra- or intraovarian factors contribute to altered follicular development during gestation, childhood, or early adulthood. Because there appears to be a genetic predisposition for PCOS, the in utero milieu may influence follicle development during gestation. Alternatively, PCO morphology is relatively common in the general population of young women. However, little is known about the regulation of ovarian and follicle development in normal postpubertal and young adolescents. It may be that during puberty, activation of ovarian growth by rising gonadotropin levels may stimulate advanced follicle development. Initiation of this process may signal and activate intraovarian factors to subsequently regulate early preantral follicle growth.
Androgen
A role for androgen excess on follicle growth and development has been suggested from ovarian morphology in hyperandrogenic women with CAH and androgen producing ovarian tumors. In particular, polycystic ovaries have been demonstrated in male to female transsexuals receiving long-term androgen treatment. Nonhuman primates treated with SC silastic capsules containing T were observed to develop enlarged ovaries and increased follicle number at all stages of growth. In situ hybridization studies revealed that AR mRNA was expressed primarily in healthy small and medium-sized antral follicles compared with large preovulatory follicles. Moreover, AR mRNA was found to colocalize with that of the FSH receptor and T treatment increased the expression of FSH receptor mRNA. The effect of androgen has also been observed in utero as prenatal administration of T to sheep and nonhuman primates resulted in phenotypic features of PCOS in postnatal animals. Notably, in nonhuman primates, the dose of T was sufficiently high to overcome the neutralizing effects of placental aromatase. In women with PCOS, mild elevation of circulating androgens during gestation would likely have little impact on the ovary. In this instance, as suggested by Dumesic and Richards, metabolic disruption during pregnancy may induce androgen overproduction by the midgestational fetal ovary to reprogram ovarian function in susceptible progeny.
Anti-Müllerian Hormone
Alternatively, there is evidence to suggest that AMH may be involved with the morphogenesis of the polycystic ovary. AMH is a product of the granulosa cell, and mRNA expression is initially detected in primary follicles. With subsequent growth, the concentration of AMH intensifies, and the greatest expression is observed in large preantral and small antral follicles ( Fig. 21.12 ). Thereafter, AMH expression declines and is not observed in late stage antral follicles, preovulatory follicles, or the corpus luteum. It has been proposed that AMH has two potential functions. First, AMH may serve to inhibit recruitment of primordial follicles into the pool of growing follicles to prevent early depletion. Second, AMH may decrease follicle sensitivity to gonadotropin stimulation to control the number of large preantral and small antral follicles that reach the preovulatory stage. It has been reported that AMH protein expression is decreased in preantral and early antral follicles of women with PCOS compared with that noted in normal follicles. This provided evidence for an accelerated entry of primordial follicles into the pool of growing follicles. By comparison, AMH mRNA and receptor gene expression were shown to be increased in granulosa cells obtained from PCOS women undergoing in vitro fertilization. Moreover, Pellatt et al. reported that AMH production was substantially greater in PCOS granulosa cells from small antral follicles than that of normal cells. These findings are consistent with clinical observations that the numbers of small antral follicles in PCOS and normal women are positively correlated with circulating levels of AMH. Thus presence of increased intraovarian AMH may limit preantral and early antral follicle growth, accounting for higher numbers of these follicles in PCOS compared with normal women.
Recently, it has been demonstrated that cultured secondary follicles from nonhuman primates exposed to AMH for 2 weeks exhibited advanced growth to the antral stage earlier than follicles untreated with AMH. Beyond 2 weeks, enhanced follicle development by AMH was not evident. Conversely, in the same study, blocking endogenous AMH activity by AMH antibody delayed follicle development to the antral stage. Notably, preantral follicle responses to AMH were variable, as growth was not advanced in some. These findings are consistent with results from other reports, including cultured human cortical tissue, in which addition of AMH promoted preantral follicle growth. Collectively, these results suggest that AMH may accelerate growth of small growing follicles but restricts maturation of antral follicles, at least in nonhuman primates. This bimodal, stage-dependent effect of AMH on follicle development does not appear to exist in mice. Whether increased local production of AMH in women with PCOS serves to advance growth of prenatal follicle while restraining the maturation of antral follicles, thus giving rise to polycystic ovarian morphology, is unknown.
Gonadotropins
The role of LH and FSH on the morphogenesis of the polycystic ovary has not been carefully examined. While growing and antral follicles are responsive to FSH, women with gonadotropin and gonadotropin receptor mutations have been shown to exhibit ample follicle development, which reflects gonadotropin-independent follicle growth. However, FSH secretion in women with this disorder is diminished and likely has little bearing on the ovarian phenotype. In contrast, LH secretion in increased in PCOS women and stimulates androgen overproduction by hyperplastic theca cells. Whether theca hyperplasia is a result of increased LH has not been shown. Terminal differentiation of granulosa cells may be induced by LH, which has led to the hypothesis that midantral follicle arrest in PCOS women may be a consequence of LH hypersecretion through premature luteinization.
Hypothalamic-Pituitary Interaction
In adult women with PCOS, LH secretion is characterized by increased pulse frequency and amplitude, elevated 24-hour mean serum concentrations, and greater responses to GnRH compared with those found in normal women ( Fig. 21.13 ). The mechanism(s) responsible for this increased release of LH are not well understood. A particular characteristic of LH secretion in PCOS is increased pulse frequency, the periodicity of which is approximately one hour. This rapid rate of LH release was not altered by ovarian suppression with GnRH agonist nor related to obesity. These observations imply that corresponding pulsatile release of hypothalamic GnRH is increased. In PCOS, the relationship between GnRH pulse frequency and gonadotrope responsiveness may be a key issue relative to inappropriate gonadotropin secretion. Previous studies in rodents have demonstrated preference for LHβ gene expression in response to rapid rates of GnRH delivery. Not only does GnRH drive the release of LH, but also, in normal women, it self-primes the pituitary to GnRH, thereby contributing to increased LH sensitivity to subsequent GnRH stimulation. Collectively, these findings have led to the suggestion that the profound abnormality of gonadotropin secretion in PCOS may be a primary consequence of increased hypothalamic GnRH activity.
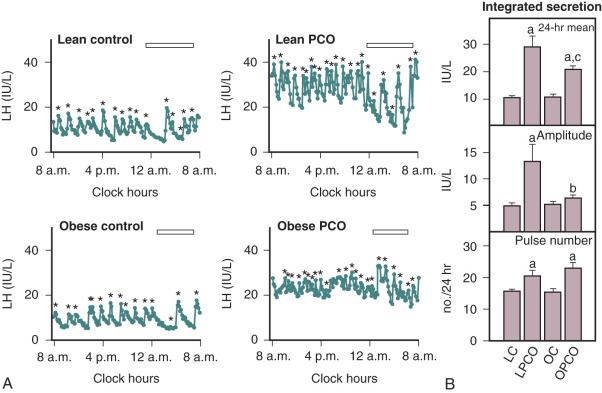
In humans, progressive increases in the frequency of GnRH stimulation have resulted in corresponding increases in the rate of LH release as well as elevated basal LH concentrations. In GnRH-deficient women, an increase in the rate of GnRH administration from every 90 minutes to every 60 minutes was not associated with corresponding increases in serum LH or changes in pulse amplitude, whereas GnRH given at 30-minute intervals resulted in elevated LH levels and reduction of pulse amplitude. While consistent with the primacy of increased hypothalamic GnRH, an LH pulse frequency of 1 hour may represent a physiological limit beyond which more frequent pulses in women do not occur. In normal ovulatory women during the late follicular phase and at midcycle, as well as in postmenopausal women, it has been documented that LH pulse frequency approximates 60 minutes. In addition, previous studies have shown that GnRH release from the medial basal hypothalamus of the fetus and adult has a periodicity of about 1 hour. Thus, in PCOS women, the tempo and to some degree the magnitude of pulsatile gonadotropin secretion are probably established by hypothalamic GnRH activity. However, beyond the requisite need for GnRH, LH responsiveness to GnRH and, accordingly, maximal increases in LH pulse amplitude are probably reliant upon other factors.
It has been suggested that the positive feedback effects of chronic estrogen secretion associated with this disorder may bring about an increase of LH either by a direct effect on gonadotrope sensitivity to GnRH or indirectly by facilitating GnRH pulse frequency. In vitro, estrogen has been shown to increase the fraction of individual gonadotropes responding to GnRH, which is consistent with amplification of LH responses to GnRH in normal women receiving estradiol benzoate. However, prolonged administration of estrone to PCOS patients failed to raise circulating levels of LH beyond baseline values or increase GnRH stimulated LH responses. Alternatively, in animals it has been demonstrated that estrogen enhances GnRH pulse frequency, and in women with PCOS serum GnRH levels are increased. That estrogen may exert an effect at the hypothalamus in PCOS is supported by the strong positive correlation of mean serum estradiol levels to GnRH pulse frequency.
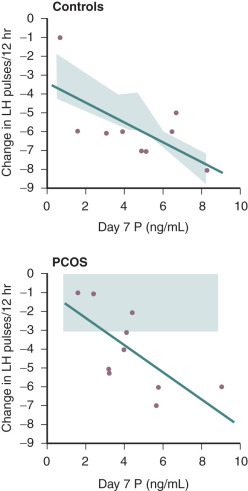
Evidence also exists that androgen may be involved in the regulation of LH secretion. Early studies had demonstrated that androgens inhibited gonadotropin secretion. Normal ovulatory women undergoing T infusion over 6 hours at high doses exhibited significant reductions in serum LH. These findings were consistent with suppression of both LH and FSH levels following 6 months of high-dose oral T administered for 6 months in female to male transsexuals. However, subsequent efforts have implicated hyperandrogenemia as a potential cause of increased LH secretion in PCOS. In vitro, it was shown that androgen administration resulted in increased GnRH pulse generator activity. Examination of LH secretion in hyperandrogenic patients with CAH revealed that mean LH levels and LH responses to GnRH were increased, which tended to normalize with the onset of treatment and corresponding lowering of androgen levels. More recently, well-designed studies have indicated that excess androgen production may have a profound, indirect influence on LH pulse frequency in women with PCOS. Progesterone administration, either alone or in combination with estrogen (oral contraceptive), suppressed mean LH and LH pulse frequency in both PCOS and normal women. The lowering of LH release was more pronounced in normal women compared with those with PCOS and suggested an effect of increased androgen production. In subsequent studies, pretreatment with an androgen blocking agent prior to physiologic estrogen and progesterone administration resulted in restoration of LH pulse frequency to that observed in normal women ( Fig. 21.14 ). These findings suggested that, in PCOS, high circulating levels of androgen prevent the negative feedback effects of estrogen and progesterone on LH pulse release as noted in earlier studies. Moreover, the physiological relevance in women underlies a rapid rate of spontaneous hypothalamic GnRH activity, which is regulated by the feedback effects of ovarian steroids in the absence of hyperandrogenemia.
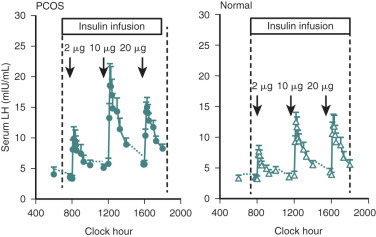
Efforts to determine an effect of insulin in PCOS women have not documented consistent alterations in LH secretion or LH release following GnRH stimulation. Reduction of hyperinsulinemia by administration of insulin-lowering drugs to PCOS patients resulted in decreased mean serum levels of androgens and LH in some cases, whereas in others an accompanying fall of LH was not found. We have explored the role of insulin on gonadotropin secretion in PCOS and normal women. Our results demonstrated that episodic gonadotropin secretion and LH responses to multidose GnRH stimulation were not altered by insulin infusion over an interval of 12 hours in both female groups. In particular, endogenous serum LH levels were unchanged prior to and immediately following initiation of the insulin clamp ( Fig. 21.15 ). These findings confirm and clarify previous studies, which have been unable to document consistent alterations in LH secretion or LH release following GnRH stimulation following insulin administration in women with PCOS. In addition, the results may explain why changes in serum LH were not observed in PCOS women treated with insulin-lowering drugs despite significant reductions in circulating androgen levels. Alternatively, interpretation of these clinical trials is potentially confounded by several factors. First, most of the patients studied were obese, and it has been shown recently that obesity is inversely correlated to LH secretion in PCOS ( Fig. 21.16 ). Second, hyperinsulinemia is positively correlated with the BMI in women with this syndrome. Third, the occurrence of ovulation in PCOS is associated with a lowering of LH levels into the normal range.
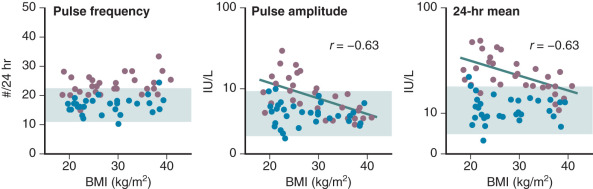
In a subsequent study, we reexamined the relationship of insulin to composite basal LH secretion and LH responses to GnRH in normal and PCOS women using rigorous analytical methods. In normal women, insulin was negatively correlated to mean LH levels. In contrast, in women with PCOS, multivariable modeling revealed that insulin had a suppressive effect on baseline LH and LH responses to GnRH dosing. These observations provided evidence for a role for insulin in modulating LH levels.
Recently, it has been reported that gonadotropin secretion, at least in part, may be regulated by AMH. In the rodent model, in vitro exposure of hypothalamic brain-slice tissue to AMH resulted in an increased firing rate of GnRH neurons. In addition, in vivo injection of AMH into the lateral ventricle of female mice produced a rapid increase of serum LH. While the action of AMH was observed in only 50% of GnRH neurons, the coexistence of elevated AMH in women with PCOS together with increased episodic LH release suggests that AMH may influence inappropriate gonadotropin secretion in this disorder.
Increased LH pulse frequency and other features of PCOS have been described in women with epileptic disorders or women treated with antiepileptic drugs. The link between epileptic and postseizure ictal states may involve stimulation of excitatory neurotransmitters, the receptors of which exist in hypothalamic nuclei that influence GnRH release. Thus the consequences of epileptic activity may result in increased GnRH activity and simulate the pattern of increased LH secretion in PCOS. In addition to altered LH secretion, polycystic ovaries and hyperandrogenism have been reported in untreated and treated women with epileptic seizures, which strengthen the possible association between PCOS and epilepsy. A mechanistic role for antiepileptic medication, including sodium valproate, in the development of excess androgen production or follicle cyst formation in treated subjects has not been elucidated.
In contrast to pituitary LH, FSH secretion in PCOS is decreased as indicated by significantly lower serum concentrations compared those found in normal women during the early follicular phase of the menstrual cycle. In addition, FSH responses to GnRH stimulation are reduced as shown in some but not all studies. The precise underlying basis for decreased FSH secretion in PCOS has not been determined, although the negative feedback effect of chronic unopposed estrogen secretion in these women has been implicated as a mechanism. The reduction in serum FSH may also reflect the activity of hypothalamic GnRH. As mentioned earlier, increased frequency of pulsatile GnRH predisposes to a preference for LHβ gene expression at the expense of the FSHβ gene.
Theca Cell Function
The most notable clinical feature of PCOS is hirsutism, which is the result of excessive androgen production. Both the ovary and adrenal glands contribute to the pool of increased circulating androgens. However, in PCOS the major serum androgens, A and T, are produced by the ovary, whereas elevated concentrations of DHEAS are derived from the adrenal glands. Within the polycystic ovary, numerous antral follicles are surrounded by hyperplastic theca cells that are the predominant site of androgen overproduction. There is firm evidence to indicate that excess ovarian androgen is driven by the abnormally increased secretion of pituitary LH acting on theca cells. Nonetheless, the rather broad range of LH secretion in PCOS suggests that other mechanisms may be instrumental in the excessive production of androgens, including increased theca cell sensitivity to LH and the presence of cogonadotropic growth factors.
In vitro studies have established that cultured theca cells from PCOS ovaries produce significantly more androgen following LH stimulation than that generated by similarly treated theca cells from normal ovaries. In these studies the rate of gonadotropin-stimulated androgen production in PCOS theca cells was greater than of normal cells, but the magnitude of response was similar in both groups, reflecting higher basal production of androgen in PCOS cells. Clinically, stimulation of ovarian androgen production by a GnRH agonist revealed significant increases in serum 17-hydroxyprogesterone (17-OHP) and androstenedione concentrations in PCOS compared with those of normal women. Differences in T responses between the two groups were not found. These results suggested an overexpression of CYP17A1 (CYP17), which regulates 17-hydroxylase and C-17-20-lyase activity. Moreover, these findings suggested that differential androgen production in PCOS may arise de novo within the theca cell. To further examine this issue, steroid responses to hCG were examined in PCOS subjects and normal subjects prior to and following administration of a GnRH agonist, which effectively suppressed and eliminated any disparity in basal LH secretion and ovarian androgen production. Serum 17-OHP responses to hCG were not abrogated by GnRH agonist treatment, providing further evidence that in PCOS the magnitude of androgen responsiveness to LH may be due to a primary abnormality of theca cell steroidogenesis.
Rosencrantz and colleagues have performed a dose-response study in PCOS women and normal women to determine theca cell androgen production following intravenous administration of multiple doses of human chorionic gonadotropin (hCG). In PCOS women 17-OHP responses to hCG increased in a dose-dependent manner and were greater than those observed in normal women ( Fig. 21.17 ). A shift in the response curve was not observed, which indicated greater theca cell responsiveness and not increased sensitivity to hCG. Notably, peak A and T responses in PCOS women occurred at a low dose of hCG, whereas maximal 17-OHP responses were observed at higher doses of hCG. In contrast, hCG-stimulated responses of A and T in normal women were not observed. These results suggested exquisite theca cell responsiveness to hCG in PCOS women compared with minimally stimulated androgen production in normal women. Studies in cultured human theca cells have demonstrated that radiolabeled pregnenolone was more rapidly metabolized to 17-OHP, pregnenolone, and DHEA, and, eventually, to androstenedione compared with that observed in normal theca cells. While 17-hydroxylation was increased in PCOS theca cells compared with normal cells, evidence of delta-4 17, 20-lyase activity was lacking in both. It was concluded that androgen biosynthesis occurred predominantly through the delta-5 pathway in PCOS as well as normal theca cells. Inefficiency of delta-4 17, 20-lyase activity may explain, at least in part, the discordant dose-responses of 17-OHP compared with those exhibited by A and T in our PCOS women. The accumulation of A and T with minimal 17-OHP production at a low dose of hCG is compatible with hyperactivity of the delta-5 steroid pathway combined with increased 3β-HSD enzyme activity, a stable property of the PCOS theca cell. At higher doses of hCG, incremental changes in 17-OHP responsiveness likely arose from combined over-expression of both 17-hydroxylase and 3β-HSD enzymes.
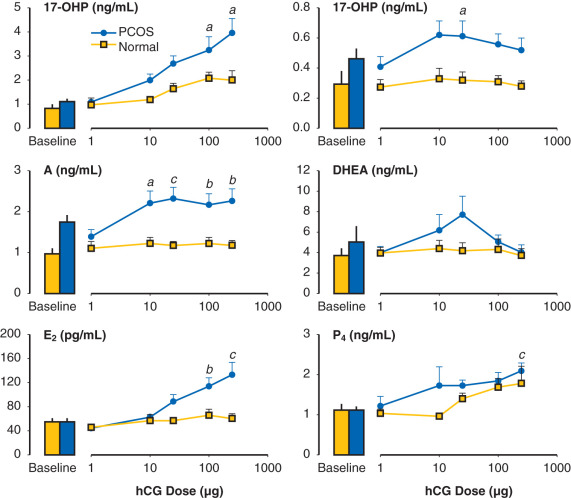
It is clear that LH is necessary to sustain circulating androgen levels. However, the extent to which endogenous LH contributes to theca cell androgen production is not completely obvious. To address this issue, we examined theca cell response to intravenous hCG stimulation in PCOS and normal women prior to and 2 days following administration of a GnRH antagonist. In both groups, antagonist treatment significantly lowered LH and moderately reduced FSH, which was accompanied by decreased basal androgen levels ( Fig. 21.18 ). Corresponding androgen responses to hCG were also lower in each group. However, the incremental fold-change for both PCOS women and normal women was not different from responses observed before agonist treatment. Our results extend the findings of other previously reported studies that endogenous gonadotropin secretion contributes to basal androgen secretion and, even in the presence of lowered gonadotropin levels, maintains ovarian androgen synthesis and responsiveness to gonadotropin stimulation. This is consistent with primary theca cell dysfunction leading to excessive ovarian androgen production in women with PCOS.
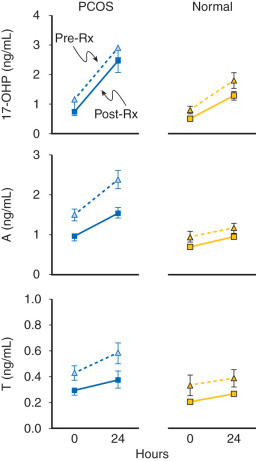
It is noteworthy that excess androgen production may also vary among individual women with PCOS as reflected by 17-OHP and androgen responses to gonadotropin stimulation. As reported by Pasquali et al., some PCOS women exhibited higher 17-OHP responsiveness to GnRH agonist administration compared with normal ovulatory controls, whereas responses in others were essentially indistinguishable from that of normal controls. It was suggested that variable responsiveness may have been an effect of hyperinsulinemia, as these PCOS subgroups demonstrated greater insulin responses during an oral glucose tolerance test. We have also shown similar variable 17-OHP and androgen responses to hCG among women with PCOS. Interestingly, serum LH levels were much greater in PCOS women with normal responses than those exhibiting high responses in whom LH concentrations were normal. This suggests that in normal responders androgen production is driven primarily by LH, whereas in high responders excess androgen more likely reflects heightened theca cell responsiveness due to a primary defect. In addition, serum AMH levels were significantly higher in normal responders compared with those of high responders, with AMH levels among PCOS women being inversely correlated with 17-OHP responses to hCG despite equivalent antral follicle numbers in each subgroup. Importantly, production of AMH and androgen responses to gonadotropin stimulation in women with PCOS appear to reflect a spectrum of ovarian activity that extends from normal to excessive. While the significance of these findings has yet to be determined, it is noteworthy that AMH has been shown in vitro to inhibit androgen production in a dose-dependent fashion in bovine theca cells. As to whether AMH and androgen production are physiologically interactive within the ovary warrants further study.
Exclusive of the direct stimulus-response dynamic of ovarian androgen production, the human theca cell is also subject to the effects of cogonadotropic growth factors, the most notable of which are insulin and IGFs. Receptors for insulin, IGF-I, and IGF-II have been localized to the theca compartment of ovaries from both normal women and PCOS patients. Accordingly, in vitro studies of normal human theca tissue have demonstrated that these growth factors are capable of independently stimulating androgen production themselves as well as enhancing androgen responses to LH. In particular, IGF-I and insulin increased T and A production in normal human theca tissue in the absence of LH. In contrast, recent studies have not shown an independent effect of insulin on CYP17 activity and androstenedione production in normal and PCOS theca tissue. However, in the presence of LH, there was ample expression of CYP17 and production of androstenedione by insulin. The cooperative signaling pathway(s) for coordinate insulin and LH action are unclear. Antibody to a downstream mediator of insulin signaling, inositolglycan, blocked androgen production by insulin but did not interfere with androgen responses to hCG. These results suggest that there are separate signaling mechanisms for insulin and LH. In another study, inhibition of PI3-kinase blocked insulin stimulated 17α-hydroxylase activity. However, selective inhibition of MAPK kinase did not decrease 17α-hydroxylase activity stimulated by forskolin or forskolin plus insulin. It was suggested that insulin stimulates both PI3-kinase and MAP kinase activities in human theca cells, but only PI3-kinase mediated the effect of insulin on forskolin stimulated 17α-hydroxylase activity.
Together, these results show that insulin enhances LH stimulated androgen production by theca cells. While insulin resistance is instrumental in defects of metabolic function, theca cells appear to be unaffected and are highly responsive to insulin in the presence of LH. Clinical evidence for insulin induced hyperandrogenemia has been demonstrated in women with PCOS following prolonged insulin infusion over 16 hours. Associated changes in gonadotropin levels were not observed. These findings are consistent with the observation that reduced hyperinsulinemia was associated with significant decreases of serum androgens without corresponding changes in LH in women with PCOS treated with insulin-lowering drugs.
Granulosa Cell Function
In PCOS, the mechanism(s) responsible for the persistence of ovulatory failure are not well understood. Earlier studies had implicated a deficiency of aromatase activity to explain the lack of follicle development, as estradiol concentrations were low in follicular fluid, and granulosa cells contained little measurable aromatase enzyme. However, subsequent in vitro studies have demonstrated that these cells are, indeed, vital and exhibit substantial capacity for steroidogenesis. We have previously shown that cultured granulosa cells obtained from women with PCOS exhibit a significantly greater rise of estradiol following FSH stimulation compared with that observed in normal granulosa cells, which indicated that the inherent capacity of these cells to respond to FSH was ample. Clinical studies have corroborated these in vitro findings. In women with PCOS the capacity for estradiol production in response to the stimulatory effects of FSH was clearly enhanced compared with that of normal women. Notably, this difference was dose-dependent and only manifested at 150 IU, as increases in circulating estradiol after 37.5 and 75 IU of FSH were similar in both PCOS and normal women ( Fig. 21.19 ). The amplification of estradiol responses beyond an apparent FSH threshold dose range indicated greater in vivo granulosa cell responsiveness in PCOS in the presence of an abundance of aromatase substrate, as profiles of circulating FSH levels at each dose were essentially identical in both groups. This heightened estradiol response may have reflected greater follicle number in the PCOS ovary compared with that of the normal ovary. This explanation is consistent with the report in which comparative estradiol responses to FSH in PCOS and normal women during ovulation induction were more likely related to the number of stimulated ovarian follicles rather than differences in the FSH threshold.
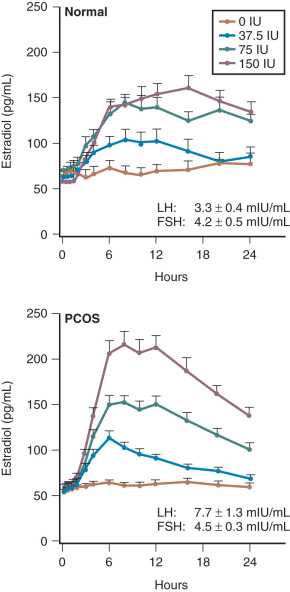
While these results may reflect increased numbers of stimulated follicles or increased granulosa cell sensitivity to FSH or both, it is evident that women with PCOS are susceptible to ovarian hyperstimulation in response to gonadotropin stimulation. Whether increased granulosa cell sensitivity may arise primarily or secondarily as a result of intraovarian or extraovarian factors is unknown. Studies have shown that in granulosa cells obtained by aspiration of follicles from unstimulated anovulatory PCOS ovaries, binding of radiolabeled FSH was significantly higher compared with that detected in cells from ovaries of ovulatory PCOS or normal women ( Fig. 21.20 ). These findings link granulosa cell sensitivity to increased FSH receptor binding in granulosa cells from PCOS ovaries and provide insight into a possible mechanism for increased estradiol responsiveness to FSH stimulation.
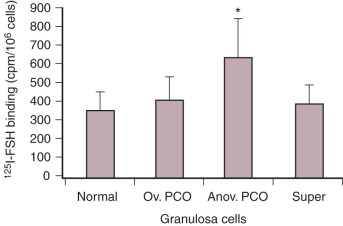
Previous studies have shown that insulin and IGFs significantly amplify steroid production in PCOS granulosa cells. In addition, the facilitatory action of insulin was likely mediated through its own receptor. However, efforts to clinically demonstrate a role for insulin on granulosa cell function in women with PCOS have been limited. Following treatment with insulin-lowering drugs, PCOS women have been shown to improve insulin sensitivity and resume ovulation, which indirectly links insulin resistance to chronic anovulation in this disorder. In studies that evaluated granulosa cell function, measurement of serum estradiol levels did not reveal any particular pattern of response in women who became ovulatory or remained anovulatory. In women undergoing weight reduction, initiation of regular menses was associated with improved insulin sensitivity as reflected by increased SHBG. In these studies, because the rate of ovulation improved during treatment, it was unclear whether an alteration of estradiol levels was a direct result of lowered circulating insulin levels or secondary to restoration of ovarian steroidogenesis. Insulin-lowering drugs, such as thiazolidinediones and metformin, have been shown to exert a direct effect on ovarian steroid enzymes. The potential effect of hyperinsulinemia on granulosa cell function has been examined by assessing estradiol responses to FSH stimulation. In women with PCOS treated with a thiazolidinedione, pioglitazone, for 5 months, estradiol production following FSH during insulin infusion was significantly increased compared with responses observed prior to treatment ( Fig. 21.21 ). Amplification of the estradiol response was associated with an improvement in insulin sensitivity that suggested in PCOS women granulosa cells may be insulin resistant. Moreover, these findings may account for the apparent paradoxical effect of insulin on granulosa cell function in that, in vitro, insulin enhances estradiol production whereas, in vivo, treatment with insulin-lowering drugs induces ovulation. Whether granulosa cells in PCOS women may be insulin resistant on a primary basis or secondarily has not been addressed.
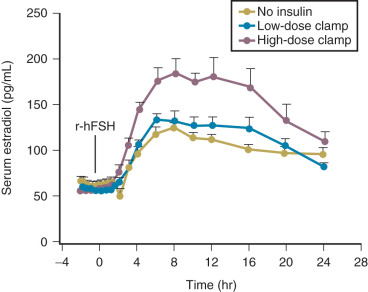
That PCOS granulosa cells may be insulin resistant, could, at least in part, account for decreased estradiol responsiveness to FSH observed in PCOS women undergoing controlled ovarian hyperstimulation. Clinical experience has indicated that this reduced follicle responsiveness may be overcome with progressive increases in the dose or duration of FSH. Protracted administration of low-dose FSH has been particularly useful as the risk of ovarian hyperstimulation syndrome is considerably diminished with this method of ovulation induction. The gradual accumulation of constant low-dose FSH over time is preferred to an escalation of the daily dose because of follicular hyperresponsiveness to FSH in PCOS women and an increased risk of ovarian hyperstimulation syndrome. These observations combined with our previous report describing greater estradiol responsiveness in PCOS women compared with normal women at a 150 IU dose of FSH suggests a bidirectional narrowing of the FSH threshold range in this disorder ( Fig. 21.22 ).
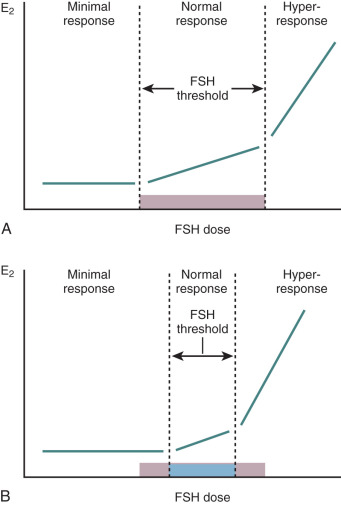
In the past it appeared that the effect of insulin on FSH stimulated follicle development may have involved the IGF system, as both IGF-I and IGF-II were shown to enhance granulosa cell responses to FSH. In PCOS granulosa cells, exposure to both IGF-I and FSH resulted in significantly greater estradiol production compared with incubation with each hormone separately. The actions of IGFs are likely mediated by their receptors, which have been identified on granulosa cells of both PCOS and normal follicles. However, despite in vitro studies in which IGF-I clearly amplified FSH induced aromatase activity, most studies have failed to detect or have shown very little of the protein in human granulosa cells. By comparison, IGF-II mRNA message has been located in all compartments of the human ovary and is strongly expressed in granulosa cells. Consistent with this observation are recent studies of human PCOS ovarian tissues that demonstrated that estrogen responses to IGF-II were significantly enhanced in granulosa cells preincubated with insulin than without insulin. These findings point out the potentially critical relationship between insulin and IGF-II in the regulation of granulosa cells function. In particular, in PCOS, excess insulin secretion may downregulate insulin receptors, which would prevent the translocation of subcellular IGF-II receptors to the plasma membrane and deprive granulosa cells of the stimulatory effect of a potent cogonadotropin.
IGFs are complexed with IGF binding proteins (IGFBPs) that regulate their bioactivity. In PCOS serum, IGFBP-1 has been shown to be decreased as a result of hyperinsulinemia, which implies an increase in the levels of free IGF-I. This notion is supported by the demonstration of increased circulating levels of free IGF-I in PCOS women. Additionally, IGFBPs may be critical in regulating IGF bioactivity at the level of the ovary. IGFBP-2 and -4 have been shown to be increased in follicle fluid derived from androgenic follicles including those of PCOS patients. IGFBP-4 protease was not detectable in these follicles. These results are in stark distinction to those found in healthy antral follicles of normal women in which IGFBP-2 and -4 were decreased and IGF-II concentrations were increased in the follicular fluid. The impact of the androgenic environment on follicular fluid IGF bioavailability has been documented in transsexuals treated with high-dose androgens. In the follicular fluid of these individuals, IGF-II levels were decreased, IGFBPs increased, and IGFBP-4 protease was nondetectable, thus accounting for decreased bioavailable IGF-II. Whether this IGF-IGFBP profile is a consequence of follicular fluid androgen levels is not clear, although the association is compelling and suggests that androgen excess in PCOS, driven in part by insulin-mediated theca cell androgen production, may be instrumental.
The potential role of the oocyte in abnormal follicle growth in women with PCOS has not been investigated. Studies have demonstrated that oocyte-derived growth factors may be important to follicle development and function. In particular, growth differentiation factor-9 (GDF-9) and bone morphogenetic protein-15 (BMP-15) appear to have major roles on folliculogenesis and female fertility. In vitro studies have shown that these genes are selectively expressed in developing oocytes during folliculogenesis. In addition, in GDF-9 deficient female mice, disruption of reproductive function has been associated with arrested follicle growth at the primary stage, decreased granulosa cell proliferation, inappropriate theca cell development, ovarian cyst formation, and infertility. We have examined mRNA expression of GDF-9 and BMP-15 in ovaries obtained from women with PCOS and found that the GDF-9 signal was decreased in oocytes throughout folliculogenesis compared with GDF-9 message in oocytes from normal ovaries. By comparison, there was no difference in BMP-15 mRNA expression between groups. These results suggest that the expression of GDF-9 is delayed in oocytes of developing PCOS follicles. Because of the apparent growing importance of GDF-9 in folliculogenesis and fertility, dysregulation of GDF-9 expression may contribute to aberrant folliculogenesis in PCOS women. Interestingly, in a subsequent study it was demonstrated that GDF-9 expression in oocytes of PCOS women undergoing ovulation induction was similar to that observed in oocytes of similarly treated normal women. That FSH stimulation may have restored GDF-9 expression in oocytes of PCOS women is consistent with well-documented inappropriate gonadotropin secretion characteristic of this disorder.
Intrafollicular Paracrine Interaction
In 2008, Wachs and coworkers reported that in PCOS women, intravenous administration of FSH resulted in significant increases in serum 17-OHP, androstenedione, and DHEA levels ( Fig. 21.23 ). By comparison, androgen levels in normal women remained unchanged after FSH stimulation. In PCOS women, serum estradiol responses were also greater than those of the normal group. These findings suggested that theca cell androgen production in women with PCOS may be, in part, regulated by granulosa cells. Both in vitro and in vivo studies conducted in animals have indicated that FSH may amplify LH-induced ovarian androgen production. In cultured human theca cells, androstenedione responses to LH in the presence of inhibin were clearly increased compared with those without inhibin. In addition, inhibin was able to negate the inhibitory effect of activin on human theca cell androgen production. Accordingly, in our study, the significant increases in ovarian androgens exhibited by PCOS women were accompanied by similar significant increments in FSH-stimulated inhibin B levels compared with those of normal women. A direct effect of inhibin B on theca cell androgen production is unlikely, as a receptor for this protein has not been identified. Rather, it has been shown that inhibin B binds to membrane bound binding protein, β-glycan, to form a complex that has high affinity for activin type II receptors. The sequestration of activin type II receptors prevents formation of the activin type II or type I receptor complex required for activin signaling and eventual inhibition of CYP17 within the theca cell.
To address whether inhibin mediates theca cell CYP17 mRNA expression, we examined ovarian theca cells in the presence or absence of granulosa cells treated with FSH, inhibin, inhibin antibody, or β-glycan antibody. CYP17 mRNA expression was dose-dependently increased by FSH, suggesting that paracrine factors from granulosa cells mediated CYP17 mRNA expression. In addition, antibodies against inhibin and β-glycan blocked the stimulatory effect of FSH on CYP17 mRNA expression. However, inhibin alone did not increase CYP17 mRNA level to the same extent. These findings suggested a role for inhibin in the regulation of theca cell Cyp17 mRNA expression; however, other paracrine factors produced by granulosa cells by virtue of FSH seem to be required.
We have also clinically investigated whether LH-induced androgen production may include a paracrine effect via granulosa cells stimulation by assessing Inhibin B responses to IV hCG in normal and PCOS women. In both groups, Inhibin B levels decreased significantly following hCG. These findings were consistent with in vitro studies in rats that revealed hCG-stimulated RAS-ERK1/2 activity inhibited FSH-stimulated granulosa cell activity, including Inhibin B production. Increased LH secretion in PCOS women may influence FSH receptor target genes in granulosa cells through activation of the RAS-ERK1/2 pathway and limit aromatase activity.
Another granulosa cell-derived growth factor, kit ligand, has also been shown to stimulate theca cell androgen production. In cultured bovine theca cells, kit ligand significantly increased androstenedione synthesis in the absence of gonadotropins. Moreover, kit ligand is stimulated by FSH and its receptor, c-kit, is increased by LH, which potentially creates a vigorous forward feedback loop for androgen production. Together, these findings provide a possible mechanism for FSH-stimulated androgen production in PCOS women. Consideration of an intrafollicular paracrine model of androgen production challenges the long-held two-cell, two-gonadotropin concept of ovarian steroidogenesis. However, documentation of this interaction in human ovarian tissue and whether inhibin or activin signaling and kit ligand or c-kit interaction is altered in theca cells of PCOS women has not been studied.
Adrenal Function
The role and significance of increased adrenal androgen production in women with PCOS remains unclear. Previous studies have not consistently shown that excess adrenal androgen contributes primarily to increased T and androstenedione (A4) production in PCOS women despite increased serum DHEAS in 20% to 30% of cases. This suggests that the mechanism for adrenal hyperandrogenemia may arise from either altered adrenal responsiveness to ACTH or abnormal adrenal stimulation by factor(s) other than ACTH. Studies to address this issue have produced varied results. Increased 17-OHP responses to ACTH, following dexamethasone, have been observed in women with PCOS and functional ovarian hyperandrogenism, which suggests dysregulation of CYP17. However, other studies have not been able to confirm these results. These latter studies did not employ dexamethasone suppression prior to ACTH stimulation, which may have accounted for the disparate results. Alternatively, separation of PCOS women based on 17-OHP responses to GnRH agonist revealed that basal androgen levels in high responders were minimally affected by dexamethasone, while the percent inhibition in normal responders was equivalent to that of normal women. This suggested that adrenal androgen production may contribute significantly to hyperandrogenemia in the normal responder group, although studies that address this notion have not been performed. In PCOS women, in vitro and in vivo studies have shown that serum 17-OHP and A4 responses to ACTH were significantly greater in the presence of hyperinsulinemia compared with those in the absence of elevated insulin levels. In addition, in individuals with PCOS, it has been demonstrated that ACTH administration during insulin infusion was associated with a significantly higher 17-hydroxypregnenolone and 17-OHP responses than those measured during saline infusion. This facilitatory effect of insulin appeared to result in a relative lowering of 17 to 20 lyase activity as indicated by higher 17-hydroxypregnenolone to DHEA and 17-OHP to A4 ratios. These findings are consistent with reports that demonstrate a reduction in serum DHEA-S in women during administration of insulin infusion or a glucose tolerance test.
Recently, we have examined 17-OHP and androgen production during a step-wise, dose-response infusion of ACTH in normal and PCOS women who had previously undergone hCG stimulation. The results revealed that in women with PCOS, androgen responses during ACTH infusion were similar to those observed in normal controls. However, A4 and DHEA responses among PCOS women were highly variable. The differing magnitudes of response suggest that adrenal androgen production is not the same in all women with PCOS and may relate to the inconsistent prevalence of increased serum DHEA-S levels reported in this disorder. Notably, androgen responses to ACTH infusion correlated significantly with those following hCG stimulation ( Fig. 21.24 ). These findings indicated a commonality of dysregulated CYP17 activities during steroidogenesis in both adrenal and ovarian tissues of PCOS women, as previously suggested. Whether adrenal hyperandrogenemia contributes to the perpetuation of PCOS has not been established. However, that an exaggerated adrenarche may be an inciting event in the pathogenesis of PCOS warrants further investigation of adrenal gland function in adolescent girls with hyperandrogenism.
Related posts:
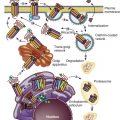 Gonadotropin Hormones and Their Receptors
Gonadotropin Hormones and Their Receptors
 Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
 Structure, Function, and Evaluation of the Female Reproductive Tract
Structure, Function, and Evaluation of the Female Reproductive Tract
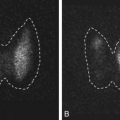 Endocrine Disturbances Affecting Reproduction
Endocrine Disturbances Affecting Reproduction
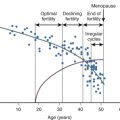 Medical Approaches to Ovarian Stimulation for Infertility
Medical Approaches to Ovarian Stimulation for Infertility
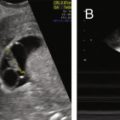 Pelvic Imaging in Reproductive Endocrinology
Pelvic Imaging in Reproductive Endocrinology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


