TABLE 101.1 CRITERIA FOR THE DIAGNOSIS OF POEMS SYNDROMEa | ||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||
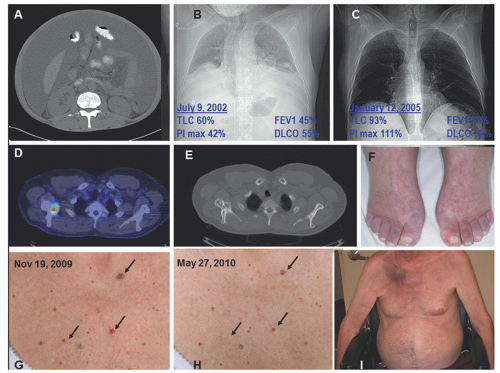 FIGURE 101.1. Classic findings of POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes) syndromes. A: Massive ascites and lipodystrophy. B: Chest radiograph and pulmonary function test results demonstrating reduced lung volumes due to neuromuscular weakness, small effusions, and reduced diffusing capacity of carbon monoxide. C: Improved chest radiograph and pulmonary function tests 2.5 years after ASCT (same patient as H). D: Fusion CT PET of mixed lytic/sclerotic lesion in right scapula. E: Bone windows of CT of mixed lytic/sclerotic lesion in right scapula. F: Hyperemia of extremities and white nails. G: Outcropping of cherry angiomata at diagnosis. H: Shrinkage and disappearance of cherry angiomata after radiation to solitary osteosclerotic lesion right femur. I: Plasmacytoma right scapula with overlying erythema as well as gynecomastia, muscle wasting, and ascites. Also present but unrelated is florid tinea corporis due to chronic steroid used for the incorrect diagnosis of CIDP. (From Dispenzieri, A. How I treat POEMS syndrome. Blood 2012;119(24):5650-5658, with permission.) |
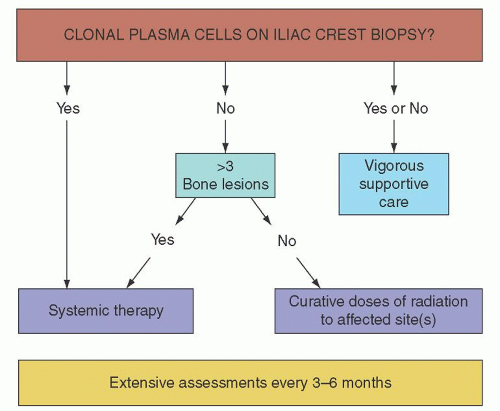 FIGURE 101.2. Algorithm for the treatment of POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes) syndrome. (From Dispenzieri, A., How I treat POEMS syndrome. Blood 2012;119(24):5650-5658, with permission.) |
TABLE 101.2 ACTIVITY OF THERAPY FOR THE TREATMENT OF POEMS SYNDROME | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
treated with lenalidomide.93 In France, nine patients, one of whom was newly diagnosed, were treated with lenalidomide and dexamethasone.94 Serious side effects were noted in three patients, with two hematologic toxicities and a cutaneous allergy. All patients evaluable for hematologic response had at least a partial hematologic response. Clinical responses, including improvement in performance status and neurologic symptoms, were documented among the 8 who had sufficient follow-up. One patient relapsed 5 months after discontinuing therapy, but responded to reintroduction of the drug. Bortezomib use has been reported in three patients.97, 98, 101 The first report is difficult to interpret, since the patient had a number of chemotherapies prior to receiving a bortezomib, doxorubicin, and dexamethasone combination. There was early evidence of improvement even before starting the bortezomib regimen. The second and third reports are more convincing: in the first, 7 cycles of bortezomib and dexamethasone resulted in patient improvement; and in the second, 18 months of cyclophosphamide, bortezomib, and dexamethasone resulted in dramatic improvements in a patient with refractory paracentesisdependent ascites. Although an anti-VEGF strategy is appealing, the results with bevacizumab have been mixed.22, 23, 24, 25, 26, 27 Five patients who had also received either radiation or alkylator during and/or predating the bevacizumab had benefit,24, 25, 26, 103 including three who had improvement but were then consolidated with high-dose chemotherapy and autologous stem cell transplant.25, 103 Three patients receiving bevacizumab died.21, 22, 27
and parvovirus B19.116, 143, 147 The association with HCV was reported in 1990 and 1991,145, 146 and the majority (42% to 100%) of patients with mixed cryoglobulinemia were found to be infected with HCV.146, 148, 149, 150, 151, 152, 153, 154, 155
TABLE 101.3 CRYOGLOBULINEMIA: CLINICAL AND EXPERIMENTAL ASSOCIATIONS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
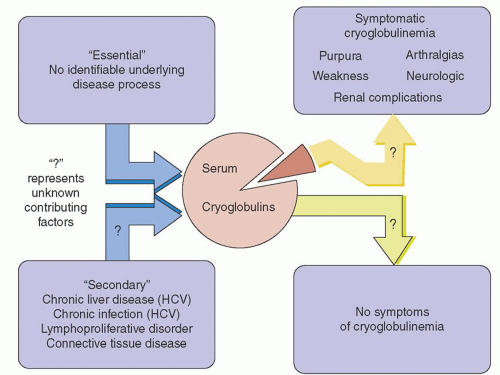 FIGURE 101.3. Relationship among underlying diseases, cryoglobulins, and symptoms of cryoglobulinemia. Question marks represent unknown contributing factors. HCV, hepatitis C virus. (Adapted from Dispenzieri A, Gertz MA. Cryoglobulinemia. In: Gertz MA, Greipp PA, eds. Handbook of Multiple Myeloma and Related Cell Disorders (Primary Amyloidosis, AL). Berlin: Springer-Verlag, 2004.) |
in regions where HCV occurs at higher frequencies (for example, southern Europe).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


