Platelet Signal Transduction
Charles S. Abrams
Lawrence F. Brass
Platelets become activated when agonists such as collagen or thrombin bind to receptors on the platelet surface, initiating intracellular signaling events that eventually lead to reorganization of the platelet cytoskeleton, platelet aggregation, and platelet granule secretion. This process can only occur when local barriers to platelet activation are exceeded or overwhelmed. Platelets are normally shielded from the connective tissue matrix within the vascular wall by a continuous barrier of endothelial cells which physically separates platelets from platelet activators in the vessel wall. When vascular injury occurs, platelets adhere to exposed collagen fibrils, forming a discontinuous carpet. In vitro, adhesion to collagen can occur without the help of accessory molecules, but in vivo, the shear forces and turbulence caused by flowing blood would strip platelets away from collagen were it not for the stabilizing effects of von Willebrand factor (vWF). The metalloprotease ADAMTS13 cleaves vWF to prevent the accumulation in plasma of ultrahigh molecular weight multimers that would otherwise cause spontaneous platelet clumping and arterial thrombosis.1,2,3,4 vWF binding to collagen via its A3 domain exposes binding sites on the vWF A1 and C1 domains for the platelet adhesion receptors, GP Ib-IX-V and αIIbβ3 (GP IIb-IIIa), respectively.5,6 Combined with the activating signals produced when collagen binds directly to the α2β1 integrin and to glycoprotein VI on the platelet surface, platelets that have adhered to collagen change their shape, spreading along the collagen fibrils, and releasing into the circulation thromboxane A2 (TxA2) and adenosine diphosphate (ADP) (FIGURE 33.1).
Released TxA2 and ADP recruit additional platelets, causing them to stick to each other and to the platelets directly adherent to collagen. The growing mound of activated platelets is eventually stabilized by a crosslinked fibrin clot, but the critical contact between adjacent platelets depends on the binding of fibrinogen or fibrin to αIIbβ3 on the platelet surface, and this occurs only after platelets have been activated. Since fibrinogen is a symmetrical molecule, it can bind to two platelets at the same time, as can fibrin. Repeated as many as 80,000 times per platelet, this allows platelets to stick to each other, a process that can be made to occur when platelets are in suspension, but should normally occur only at a site of vascular injury. Like collagen, soluble platelet agonists cause platelets to change their shape, losing the discoid appearance characteristic of resting platelets, and transform them into an irregular sphere with pseudopodia. Underlying this shape change is rapid reorganization of the platelet cytoskeleton as actin filaments are uncapped, severed, and rebuilt in response to intracellular signals that include a rise in the cytosolic Ca2+ concentration and the sequential activation of low molecular weight GTP-binding proteins such as Rac and Rho.
Collagen, ADP, and TxA2 are not alone in their ability to activate platelets. Vascular injury and inflammation expose tissue factor as well as collagen, and formation of the tissue factor/factor VIIa complex leads to the local generation of thrombin from prothrombin. Thrombin is a potent agonist, activating platelets at concentrations in the pM range by interacting with receptors on the platelet surface. Activated platelets facilitate this process by providing procoagulant phospholipids that accelerate thrombin generation. As a result, platelet activation and fibrin deposition are intimately linked, maximizing the growth and strength of the hemostatic plug. Human platelets express two G-protein-coupled receptors (GPCRs) that can be activated by thrombin. Cleavage of these receptors accounts for the long-established observation that only proteolytically active thrombin can activate platelets.
The process of transforming αIIbβ3 on the platelet surface into a competent receptor for fibrinogen is one of the most fascinating aspects of platelet biology.7 It is also the final common pathway in platelet responses to most agonists, making it a frequent target for drug development. Stated succinctly, the central issue is this: although circulating platelets are surrounded by plasma proteins, they normally do not bind fibrinogen to their surface and stick to each other unless they have been activated. The reasons for this are multiple, but are ultimately due to the inability of fibrinogen or fibrin to bind to the resting conformation of αIIbβ3. Platelet activation alters the conformation of αIIbβ3 and exposes the fibrinogen-binding site. Normally, this should occur only at sites of vascular injury. However, even in the absence of substantial vascular injury, it is likely that the buffeting that platelets withstand as they move through the circulation, perhaps encountering low concentrations of thrombin or fractured atherosclerotic plaques, pushes them toward inappropriate activation.
Working against this tendency are a number of internal and external controls that dampen intracellular signals that would otherwise promote platelet activation. In addition to providing a physical barrier and a source of vWF, endothelial cells release PGI2 and NO, two molecules that globally depress intracellular signaling in platelets by raising cyclic AMP (cAMP) and cGMP levels.8,9,10,11,12,13,14 The ability of cyclic nucleotides to inhibit platelet activation has been exploited in the development of antiplatelet agents such as dipyridamole (Persantine), which raises cAMP levels inside platelets by inhibiting cAMP phosphodiesterase.15 Cyclic GMP also inhibits cAMP phosphodiesterase in platelets, contributing to the rise of cAMP levels. In turn, cAMP activates protein kinase A, which phosphorylates multiple platelet proteins including the β chain of GP Ib,16,17 actin-binding protein (filamin),18 myosin light chain,19,20 vasodilator-stimulated phosphoprotein (VASP),21,22 and Rap1B.23 The net effect is a generalized inhibition of platelet activation. The importance of PGI2
and NO as barriers to platelet activation is indicated not only by the effectiveness of molecules that mimic PGI2 as antiplatelet agents, but also by the prothrombotic effects in mice of deleting the gene encoding the platelet PGI2 receptor (IP)24 and by the decrease in basal cAMP levels found in IP-/- platelets.25 In addition to releasing PGI2 and NO, some endothelial cells express the ecto-ADPase CD39 on their luminal surface. CD39 can hydrolyze the small quantities of ADP released from damaged red cells and activated platelets, preventing the ADP from activating additional platelets.26,27 Other barriers to unwarranted platelet activation include the diluting effects of continued blood flow, the presence of natural anticoagulants that limit thrombin formation, the short half-life of the platelet-derived agonist, TxA2, and a variety of mechanisms that limit the duration of signaling by G proteins and GPCRs. This includes receptor desensitization, receptor internalization, and regulators of G-protein signaling (RGS) proteins. Collectively, these provide a threshold that helps to prevent platelet activation at inappropriate times and places.
and NO as barriers to platelet activation is indicated not only by the effectiveness of molecules that mimic PGI2 as antiplatelet agents, but also by the prothrombotic effects in mice of deleting the gene encoding the platelet PGI2 receptor (IP)24 and by the decrease in basal cAMP levels found in IP-/- platelets.25 In addition to releasing PGI2 and NO, some endothelial cells express the ecto-ADPase CD39 on their luminal surface. CD39 can hydrolyze the small quantities of ADP released from damaged red cells and activated platelets, preventing the ADP from activating additional platelets.26,27 Other barriers to unwarranted platelet activation include the diluting effects of continued blood flow, the presence of natural anticoagulants that limit thrombin formation, the short half-life of the platelet-derived agonist, TxA2, and a variety of mechanisms that limit the duration of signaling by G proteins and GPCRs. This includes receptor desensitization, receptor internalization, and regulators of G-protein signaling (RGS) proteins. Collectively, these provide a threshold that helps to prevent platelet activation at inappropriate times and places.
 FIGURE 33.1 Stages in platelet plug formation. Prior to vascular injury, platelets are restrained from activation by the inability of plasma vWF to bind spontaneously to the platelet surface. The development of the platelet plug can be initiated by the exposure of collagen in the vessel wall or by the local generation of thrombin (or both). Rolling platelets adhere and spread on the collagen/vWF matrix, forming a monolayer of activated platelets that acts as a surface for subsequent recruitment of platelets by thrombin, ADP, and TxA2. |
THREE STAGES OF PLATELET PLUG FORMATION
One might consider the platelet surface as being crowded with receptors that support one or more of the phases of platelet plug formation. In fact, approximately half of the platelet’s surface is occupied by αIIbβ3. Receptors that are directly involved in binding to adhesive proteins such as collagen, vWF, and fibrinogen are present in the greatest numbers. After recruitment of additional pools of receptors that are initially within the surface-connecting membrane system or in the membranes of α granules, there are approximately 80,000 copies of αIIbβ3 and 15,000 copies of GP Ib on the surface of human platelets (these receptors are also discussed in Chapters 26A, 26B, and 31). In contrast, receptors that primarily serve as signaling response elements for platelet agonists typically range from a few hundred to a few thousand per platelet (Table 33.1). Although these numbers are not large in absolute terms, when placed in the context of the relatively small size of human platelets, the density of these receptors is high. In cells other than platelets, it has been shown that some signaling molecules tend to be concentrated within or near cholesterolenriched microdomains within the plasma membrane.28 There is growing evidence that this occurs in platelets as well, increasing the efficiency of platelet activation.29,30,31
The platelet monolayer that forms following the exposure of collagen and vWF initiates platelet plug formation but is insufficient to prevent bleeding. The extension or recruitment phase of platelet plug formation occurs when additional activated platelets accumulate on top of the monolayer.32,33 Recruitment is mediated by the local accumulation of molecules that are released from platelets, such as ADP and TxA2. Contacts between platelets are maintained by a variety of molecular interactions, of which
the best described, and most critical, is binding of fibrinogen and vWF to activated αIIbβ3. Thrombin continues to participate in this phase of platelet activation and, in turn, activated platelets provide a procoagulant surface on which more thrombin can be generated via assembly of the tenase and prothrombinase coagulation complexes. Circulating or locally secreted epinephrine causes vasoconstriction, but also potentiates the effects of other platelet agonists. In contrast to platelet activation by collagen, platelet activation by most of the agonists involved in extension of the platelet plug is very rapid, with some responses occurring within a fraction of a second. Teleologically, this makes sense since circulating platelets would not be expected to linger at a wound site long enough for a slower process to occur. Videos of thrombus formation at sites of focal vascular injury show that initially most platelets move by the site of injury at speeds too rapid for activation to occur.32 Tethering to vWF-decorated collagen or to previously deposited platelets allows new platelets to remain in place long enough to become activated.
the best described, and most critical, is binding of fibrinogen and vWF to activated αIIbβ3. Thrombin continues to participate in this phase of platelet activation and, in turn, activated platelets provide a procoagulant surface on which more thrombin can be generated via assembly of the tenase and prothrombinase coagulation complexes. Circulating or locally secreted epinephrine causes vasoconstriction, but also potentiates the effects of other platelet agonists. In contrast to platelet activation by collagen, platelet activation by most of the agonists involved in extension of the platelet plug is very rapid, with some responses occurring within a fraction of a second. Teleologically, this makes sense since circulating platelets would not be expected to linger at a wound site long enough for a slower process to occur. Videos of thrombus formation at sites of focal vascular injury show that initially most platelets move by the site of injury at speeds too rapid for activation to occur.32 Tethering to vWF-decorated collagen or to previously deposited platelets allows new platelets to remain in place long enough to become activated.
Table 33.1 GPCRs expressed on human platelets | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Formation of the platelet plug following vascular injury can be thought of as involving three stages: adhesion, activation, and secretion (FIGURE 33.1). Adhesion begins with the rolling, arrest, and activation of moving platelets by the collagen/vWF complex to form a platelet monolayer. Activation refers to the recruitment of additional platelets through the stimulation by fast-acting agonists such as thrombin, ADP, and TxA2, all three of which activate platelets via GPCRs on the platelet surface. Secretion of additional agonists and adhesive ligands perpetuates these events by recruiting additional platelets to participate in the formation of the platelet plug. These definitions are arbitrary, since the adhesion of platelets to collagen and vWF also initiates some of the early stages of activation. In addition to these functionally defined stages in platelet plug formation, stable contacts between platelets make possible an additional wave of intracellular signaling that reinforces platelet activation. This “contact dependent” signaling is mediated in part by adhesion molecules such as αIIbβ3 and in part by platelet surface ligands such as ephrinB1 and sema4D that bind to receptors on the surface of adjacent platelets.34
PLATELET ADHESION
The arrest and eventual activation of moving platelets by collagen is accomplished by receptors on the platelet surface that either bind directly to collagen (GP VI and the integrin α2β1) or bind indirectly to collagen via vWF (the GP Ib/IX/V complex and the integrin αIIbβ3). The integrins require prior activation via intracellular “inside-out” signaling to engage collagen and vWF, after which they can contribute further platelet activation by generating outside-in signals. The presence of collagen and vWF-binding sites on GP VI and GP Ibα allows platelets to slow down long enough to become activated and fully adherent. Platelet activation by collagen is summarized in FIGURE 33.2. Intracellular signaling in response to collagen is mediated by at least three receptors: GP VI, GP Ib, and, after integrin activation has occurred, α2β1. The structure of GP VI places it in the immunoglobulin domain superfamily. Its ability to generate signals rests primarily on its constitutive association with a second molecule, the Fc receptor γ-chain.35 Platelets from mice that lack the γ-chain36 have impaired responses to collagen. So do platelets from humans,37,38 but possibly not mice,39 with reduced expression of α2β1. Using blocking antibodies or a depletion strategy, Nieswandt et al.39 concluded that GP VI is required for platelet responses to collagen.
Signaling through GP VI can be studied in isolation with the snake venom protein, convulxin, or with synthetic “collagenrelated” peptides. According to current models, collagen causes clustering of GP VI and its associated Fc receptor γ-chain. This promotes phosphorylation of the γ-chain by nonreceptor tyrosine kinases in the Src family, creating an immunoreceptor tyrosine-based activation motif recognized by the tandem SH2 domains of Syk. Association of Syk with the GP VI/γ-chain complex activates Syk and leads to the phosphorylation and activation of phospholipase C (PLC) γ2. Loss of Syk impairs collagen responses.36 PLCγ PLCg2 hydrolyzes membrane PI-4,5-P2 to form the second messengers 1,4,5-IP3 and 1,2-diacylglycerol (DAG). IP3 opens Ca2+ channels in the platelet-dense tubular system (DTS), raising the cytosolic Ca2+ concentration and triggering Ca2+ influx across the platelet plasma membrane that is initiated when depletion of the DTS Ca2+ pool triggers a conformational change in the DTS membrane protein, STIM1, allowing it to bind to Orai1 in the plasma membrane.40 DAG and Ca2+ activate the more common protein kinase C (PKC) isoforms that are expressed in platelets, making possible the regulatory serine/threonine phosphorylation events that are needed for platelet activation. At least one of those phosphorylation events regulates the exposure of fibrinogen-binding sites on αIIbβ3, but the full mechanism for activating αIIbβ3 remains obscure. Events
triggered by GP VI lead to activation of α2β1 as well as αIIbβ3. Engagement of the two integrins with their respective ligands initiates a further round of signaling that features some of the same molecules that are downstream of GP VI: Syk, SLP-76, and PLCγ2.41 This is a theme that repeats itself during platelet activation: different agonists with disparate receptors trigger signaling events that involve a common set of intermediates. Outside-in signaling is discussed at greater length in the last section of this chapter.
triggered by GP VI lead to activation of α2β1 as well as αIIbβ3. Engagement of the two integrins with their respective ligands initiates a further round of signaling that features some of the same molecules that are downstream of GP VI: Syk, SLP-76, and PLCγ2.41 This is a theme that repeats itself during platelet activation: different agonists with disparate receptors trigger signaling events that involve a common set of intermediates. Outside-in signaling is discussed at greater length in the last section of this chapter.
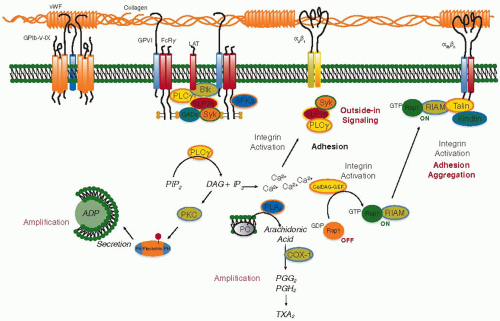 FIGURE 33.2 Platelet activation by collagen. Platelets use several different molecular complexes to support platelet activation by collagen. These include (a) vWF-mediated binding of collagen to the GPIb-IX-V complex and integrin αIIbβ3, (b) a direct interaction between collagen and both the integrin α2β1 and the GPVI/FcRγ-chain complex. Clustering of GPVI results in the phosphorylation of tyrosine residues in the FcRγcytoplasmic domain, followed by the binding and activation of the tyrosine kinase, Syk. One consequence of Syk activation is the phosphorylation and activation of PLCγ, leading to phosphoinositide hydrolysis, secretion of ADP, and the production and release of TXA2. COX-1, cyclooxygenase 1; DAG, diacylglycerol; GP, glycoprotein; PG, prostaglandin; PIP2, phosphatidylinositol-4,5-bisphosphate; PKC, protein kinase C; PLA2, phospholipase A2; PLCγ, phospholipase Cγ; TXA2, thromboxane A2; vWF, von Willebrand factor. |
Finally, GP Ib serves both as an anchor point for the platelet cytoskeleton and as the focus for assembling signaling complexes in platelets that have become bound to vWF. The cytoplasmic domain of GP Ibα can bind to filamin and to the scaffolding protein, 14-3-3ζ. Src becomes associated with GP Ib following vWF binding. The p85 subunit of PI3-kinase appears to mediate this association. In turn, Syk is activated. Thus, signaling through GP Ib takes advantage of some of the same signaling molecules that are involved in GP VI- and integrin-dependent responses to collagen, with the same potential for causing protein tyrosine phosphorylation, PI-4,5-P2 hydrolysis, and recruitment of cytosolic proteins whose Pleckstrin homology (PH) domains can support associations with 3-phosphorylated phosphoinositides.42,43,44,45,46,47,48,49
PLATELET ACTIVATION BY SOLUBLE AGONISTS
The agonists involved in extension of the platelet plug typically cause platelet activation via GPCRs on the platelet surface. GPCRs are well suited for this task in a number of respects. First, most of these receptors bind ligands with high affinity. Second, GPCRs comprise a single subunit that does not require oligomerization (although recent studies have shown that some GPCRs exist as homo- or heterodimers even in the absence of their ligands).50,51,52 Third, GPCRs are constitutively associated with G proteins, eliminating the time that would otherwise be required to recruit them into complexes.50 Fourth, because they act as guanine nucleotide exchange factors, each occupied receptor can activate multiple G proteins and, in some cases, more than one class of G proteins. This allows amplification of a signal that might begin with a relatively small number of receptors. It also enables some receptors to signal via more than one
effector pathway. Finally, because several generic mechanisms exist that can limit the activation of GPCRs, platelet activation can be limited, a property that may be useful when platelet activation is inappropriate and needs to be contained.34
effector pathway. Finally, because several generic mechanisms exist that can limit the activation of GPCRs, platelet activation can be limited, a property that may be useful when platelet activation is inappropriate and needs to be contained.34
GPCRs comprise a single polypeptide chain with seven transmembrane domains, an extracellular N-terminus, and an intracellular C-terminus. Binding sites for agonists can involve the N-terminus, the extracellular loops, or a pocket formed by the transmembrane domains.50 The G proteins that act as mediators for these receptors are heterotrimers comprised of α, β, and γ subunits. Within this complex, the β subunit forms a propellerlike structure that is tightly associated with the much smaller γ subunit. The α subunit contains a guanine nucleotide-binding site that is normally occupied by GDP. Receptor activation causes the exchange of GTP for GDP, altering the conformation of the α subunit and exposing sites on both Gα and Gβγ for interactions with downstream effectors.53,54,55 Hydrolysis of the GTP by the intrinsic GTPase activity of the α subunit restores the resting conformation of the heterotrimer, preparing it to undergo another round of activation and signaling.56 A family of GTPase activating proteins known as RGS proteins help to accelerate the hydrolysis of GTP by Gα.57 Prenylation of the γ chain and acylation of the α subunit helps to anchor the complete heterotrimer to the plasma membrane and continues to anchor Gα-GTP and Gβγ to the membrane when they dissociate from each other to expose effector binding sites. Platelets express 10 forms of Gα (Table 33.2). Although there is little information about which types of Gβ and Gγ are expressed in platelets, the forms of Gα identified have included at least one Gsα family member, four Giα family members, three Gqα family members, and two G12α family members. The roles of each have been defined through knockout studies in mice.58 Based on the available evidence, the abundance of G-protein types in platelets appears to be necessary to support the actions of multiple dissimilar platelet agonists.25
Table 33.2 G-proteins in platelets | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
GPCRs and Platelets
The GPCRs that respond to platelet agonists differ in their potency and their preferences for intracellular effector pathways. Some, such as the receptors for thrombin (PAR1 and PAR4), TxA2 (TP), and ADP (P2Y1), cause phosphoinositide hydrolysis and raise the cytosolic Ca2+ concentration by activating Gq (FIGURE 33.3).59 Others, such as the P2Y12 receptor for ADP and the α2A-adrenergic receptor for epinephrine, are coupled by Gi2 and Gz, respectively, to the inhibition of adenylyl cyclase and the activation of PI3-kinase and Rap1B.25,60,61,62 Optimal platelet activation via GPCRs is thought to require activation of both a Gq-coupled receptor and a Gi-coupled receptor.63 The ability of the four Gi family members that are expressed in platelets to inhibit adenylyl cyclase, thereby decreasing cAMP formation, is most relevant when PGI2 secreted by endothelial cells has inhibited platelet activation by raising platelet cAMP levels.25 Otherwise, it is likely that other Gi effectors account for the required Gi-coupled receptor activation during platelet activation. The biological relevance of the Gi2-coupled P2Y12 receptor for ADP is supported by the phenotypes of Gi2 and P2Y12 knockout mice and by the efficacy of P2Y12 antagonists such as clopidogrel and prasugrel as antiplatelet agents.25,62,64,65,66 Thrombin and TxA2 receptors can also cause the rearrangement of the actin cytoskeleton that underlies platelet shape change by coupling to guanine nucleotide exchange factors for Rho via G12 and G13.67 In cells other than platelets, the link from G12 family members to Rho activation is provided by proteins with RGS
domains (for binding to Gα) and DBL-PH domains (for causing guanine nucleotide exchange on Rho family members)— proteins such as p115 RhoGEF, LARG, and PDZ-RhoGEF.68 Which, if any, of these proteins plays a role in platelets remains to be determined.
domains (for binding to Gα) and DBL-PH domains (for causing guanine nucleotide exchange on Rho family members)— proteins such as p115 RhoGEF, LARG, and PDZ-RhoGEF.68 Which, if any, of these proteins plays a role in platelets remains to be determined.
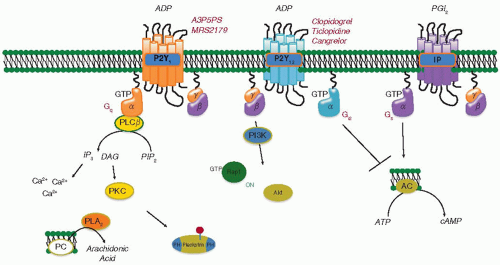 FIGURE 33.3 Platelet activation by ADP. Two receptors that can be activated by ADP have been identified in platelets. P2Y1 and P2Y12, which are coupled to different G proteins and therefore mediate different responses. Gq couples P2Y1 receptors to the activation of PLCβ. The Gi family member, Gi2, couples P2Y12 to the inhibition of cAMP formation (via Gi2α) and to effector pathways that include PI3K and Rap1B (via Gβγ). |
Platelet responses to agonists such as thrombin, ADP, TxA2, and epinephrine can be mediated by a single class of receptors coupled to different families of G proteins, by multiple classes of receptors each coupled to a single family of G proteins, or by the cumulative effect of two or more agonists, each of which evokes only a subset of the necessary G-protein-mediated responses.69,70 The next section illustrates how this is thought to occur for several essential agonists, beginning with thrombin.
Platelet Activation by Thrombin
Thrombin activates platelets at concentrations as low as 0.1 nM (˜ 0.01U/mL). Within seconds, the cytosolic Ca2+ concentration increases 10-fold to approximately 1 µM, triggering downstream Ca2+-dependent events, including the activation of phospholipase A2. Of these responses, platelet aggregation, phosphoinositide hydrolysis, and the increase in cytosolic Ca2+, but not platelet shape change, are absent in platelets from mice lacking Gqα.59 Thrombin also activates Rac and Rho in platelets, leading to rearrangement of the actin cytoskeleton and shape change—presumably mediated by a combination of effector pathways coupled to G12 and Gq activation. Finally, thrombin is able to inhibit adenylyl cyclase activity in human platelets, either directly via a Gi family member coupled to a thrombin receptor71 or indirectly via released ADP.72
Thrombin activates platelets via GPCRs of the protease-activated receptor (PAR) family, the first member of which, PAR1, was identified in the Coughlin73 and Pouysségur74 laboratories (reviewed at greater length in reference).75 Prior work had shown that platelet responses to thrombin require the enzyme’s proteolytic activity and are mediated by G proteins. Binding studies had identified high-affinity interactions with several sites on the platelet surface, including GP Ibα, but efforts to establish any of these as a receptor in the signaling sense were not entirely successful. Substrates for thrombin were identified on the platelet surface, including GP V. However, cleavage of GP V did not appear to be required for platelet activation by thrombin.
Four PAR family members have been identified, three of which (PAR1, PAR3, and PAR4) are thrombin receptors. Studies on PAR1 established a paradigm that applies to some of the other family members as well (FIGURE 33.4). PAR1 is normally activated when thrombin binds to the receptor N-terminus, cleaves it, and exposes a new N-terminus that serves as a tethered ligand. Given sufficient substrate recognition, proteases other than thrombin can also activate PAR1 or, by cleaving in the “wrong” place, render the receptor unresponsive to a subsequent addition of thrombin.76 The contact sites for the newly exposed tethered ligand have been localized to the membraneproximal region of the N-terminus and parts of the second extracellular loop.77,78,79 Because the ligand is not free to diffuse away from the receptor, it is thought to effectively present a high local concentration at the receptor and perhaps maintain signaling longer than would otherwise occur. Peptides corresponding to the tethered ligand domain (SFLLRN) can also
activate PAR1, mimicking the effect of thrombin.73,80 The same is true for PAR2 and PAR4, but not PAR3.
activate PAR1, mimicking the effect of thrombin.73,80 The same is true for PAR2 and PAR4, but not PAR3.
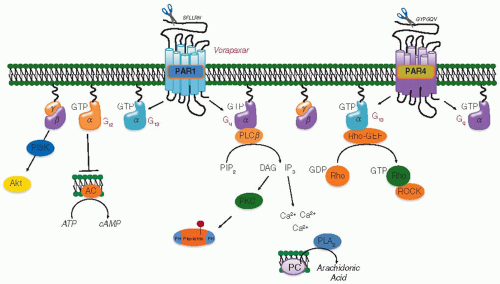 FIGURE 33.4 Platelet activation by thrombin. Platelet responses to thrombin are mediated by members of the PAR family. Human platelets express PAR1 and PAR4, which collectively are coupled to Gq– and G13-mediated effector pathways. PAR1 can also couple to Gi2, but Gi-dependent signaling initiated by thrombin gets an additional boost from secreted ADP through P2Y12 receptors. PAR1 (and presumably PAR4) are also subject to cleavage by proteases other than thrombin. Cleavage in the N-terminus by these proteases can activate PAR1 by exposing the tethered ligand domain or disable it by cleaving it elsewhere in the N-terminus that does not expose a competent tethered ligand. |
PAR1 was identified by expression cloning. The second PAR family member to be identified, PAR2, was cloned serendipitously. PAR2, expressed by endothelial cells, but not by platelets, can be cleaved and activated by trypsin, tryptase, TF/VIIa, and by factor Xa,81,82 but not by thrombin.83,84 The third family member, PAR3, was identified after gene ablation studies showed that platelets from mice lacking PAR1 were still responsive to thrombin.85 PAR3 is a major regulator of thrombin responses in mouse platelets, but appears to do so solely by facilitating cleavage of PAR4.86,87 PAR4 is expressed on human and mouse platelets and accounts for the ability of platelets from PAR3 knockout mice to respond to thrombin.88,89 Simultaneous inhibition of human PAR1 and PAR4 with blocking antibodies or small-molecule antagonists completely abolishes platelet responses to thrombin,90 as does deletion of the gene encoding PAR4 in mice.91 Thus, the four PAR family members have some features in common, but there are differences among them as well. PAR1 and PAR3 have similar thrombin dose/response curves, while PAR4 requires 10- to 100-fold higher concentrations of thrombin, apparently because it lacks the hirudin-like sequences that interact with thrombin’s anion-binding exosite and facilitate receptor cleavage.87,88,89,92 Loss of PAR3 expression on mouse platelets shifts the thrombin dose/response curve to the right,88 whereas loss of PAR4 abolishes thrombin responsiveness.91
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


