Platelet Prostanoid Metabolism
Tilo Grosser
Garret A. Fitzgerald
The prostanoids, which include prostaglandins (PGs) and thromboxane (TX) A2, are a series of lipid hormones that act on specific G-protein-coupled receptors expressed at, or close to, their site of formation. They are formed by the sequential action of the PGG/H synthases, commonly known as cyclooxygenases (COXs), and specific prostanoid synthases (figure 34.1).1 TXA2 is synthesized by COX-1 in activated platelets and released as a local signal that amplifies activation and recruits additional platelets to the site of clot formation.2,3 It is also a potent vasoconstrictor and stimulates mitogenesis and the proliferative response to vascular injury.4,5,6,7 Patients deficient in TXA2 formation or signaling have a mild bleeding disorder.8,9,10,11,12,13 Suppression of TXA2 formation by low-dose aspirin, which inhibits platelet COX, is sufficient to explain the reduction of the incidence of myocardial infarction and stroke by about 25% in patients who have had a previous arterial thrombotic event.14,15,16,17 Conversely, the defect in platelet function caused by aspirin can also lead to bleeding complications—primarily gastrointestinal and less frequently intracranial hemorrhage.
The development of nonsteroidal anti-inflammatory drugs (NSAIDs) targeted specifically at inhibition of COX-2 has emphasized the importance of PGs in the physiologic restraint of thrombotic processes. All COX-2 selective NSAIDs approved by the U.S. Food and Drug Administration were shown in placebo controlled trials to cause thrombotic complications—myocardial infarction, stroke, and venous thrombosis. The underlying molecular mechanism—inhibition of COX-2-dependent formation of prostacyclin (PGI2) and other PGs in the vasculature, the heart, and the kidney—has been elucidated in more detail than that of any other adverse drug effect.18
PGI2 is the major platelet inhibitory prostanoid. It also elicits vasodilation close to the site of formation and has antimitogenic and antiatherogenic properties.19 Augmented PGI2 biosynthesis in syndromes of platelet activation serves to constrain the effects of stimuli to platelet activation and vasoconstrictors.7 The increased incidence of myocardial infarction and stroke in patients receiving COX-2 selective NSAIDs, which is explained by inhibition of COX-2-dependent PGI2 formation, supports this concept.18,20
The role in platelet function of other prostanoids is less established. PGD2 produced cooperatively by platelet COX and plasma PGD synthase may afford a mechanism of feedback inhibition of aggregation.21,22,23 High concentrations of PGE2 may inhibit platelet function,24 while lower concentrations activate platelets in vitro.25 Deletion of the enzyme primarily mediating inflammatory PGE2 formation did not affect thrombogenesis in mice.26
PROSTANOID SYNTHESIS
Arachidonic acid, the precursor of a complex array of more than 20 carbon lipid mediators including the prostanoids, is metabolized by three major groups of enzymes, the COXs, lipoxygenases, and epoxygenases, to form the prostanoids, the leukotrienes, and the epoxyeicosatrienoic acids, respectively.27 A fourth group of arachidonic acid-derived molecules, the isoeicosanoids, is formed by nonenzymatic peroxidation. Arachidonic acid itself is released from membrane stores by the enzymatic activity of phospholipase (PL) A2. Several PLA2 enzymes that hydrolyze arachidonic acid from the sn-2 position of membrane glycerophospholipids have been identified in vertebrates.28 Free arachidonic acid is either available for metabolism into one of the 20 carbon lipid mediators, or it can be reacylated in the so-called Lands cycle, in which it is converted to arachidonoyl-CoA by arachidonoyl-CoA synthetases and reincorporated into phospholipids by CoA-dependent acyltransferases.29
Many platelet agonists initiate TXA2 generation by activating Group (G) IVA PLA2, the main isoform expressed in the platelet cytosol, with a calcium signal.30 GIVA PLA2 possesses a regulative C2 domain that is thought to mediate its translocation to the phospholipid membrane in response to increments in intracellular calcium concentrations.31 Other activation pathways, including von Willebrand factor-initiated adhesion of platelets through the GPIb-V-IX complex, are thought to involve mitogen-activated protein kinase-mediated phosphorylation of GIVA PLA2 serine-505.32,33,34,35 Vascular wall cells express multiple PLA2 isoforms to release arachidonic acid for prostanoid formation, including calcium-dependent and calcium-independent mechanisms, and in inflammatory scenarios, such as atherosclerotic lesions, also secretory PLA2s.36,37
COX-dependent metabolism of arachidonic acid results in the formation of the prostanoids. Two evolutionary conserved38 COX isoforms, COX-1 and COX-2,39 convert arachidonic acid to PGH2. COX-1 expression is relatively constant in most cells. It is the only isoform expressed in mature platelets.40 Megakaryocytes, and immature platelets released in clinical conditions of accelerated platelet turnover, also express COX-2,41,42 although its role in platelet development and function has yet to be elucidated.43
COX-2, a member of the inducible immediate-early gene family, is highly regulated in various contexts such as inflammation, pain signaling, fever, renal salt handling, and cell proliferation and differentiation. Its promoter contains several transcriptional regulatory elements including cyclic AMP response elements, nuclear factor kappa B sites, AP-2 sites, an E-box, a TATA box, an SP1 site, and a CAAT enhancer binding protein site.44 COX-2 expression is also regulated at the
posttranscriptional level by RNA-binding proteins and miR-NAs forming RNA regulons.45,46 While these influences on RNA stabilization, transport, and translation result in highly tunable gene expression, COX-2 is not only important during pathologic processes. It is developmentally expressed and has key functions in uninflamed tissues including the vasculature, kidney, gastric epithelium, spinal cord, and brain.38
posttranscriptional level by RNA-binding proteins and miR-NAs forming RNA regulons.45,46 While these influences on RNA stabilization, transport, and translation result in highly tunable gene expression, COX-2 is not only important during pathologic processes. It is developmentally expressed and has key functions in uninflamed tissues including the vasculature, kidney, gastric epithelium, spinal cord, and brain.38
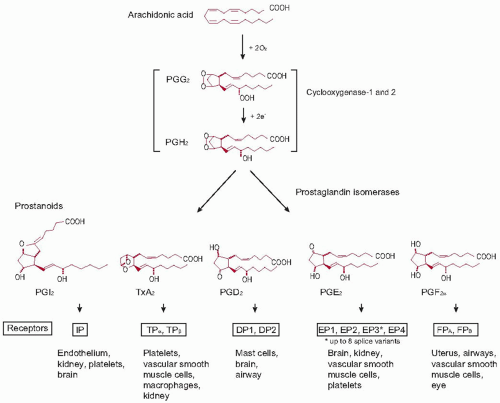 FIGURE 34.1 The cyclooxygenase pathway |
The cyclooxygenase enzymes function as membraneanchored dimers.47 The two monomers act synergistically; inactivation of one monomer is sufficient to inhibit enzyme function.47,48 Enzymatic PG synthesis involves both a COX and a peroxidase reaction. The COX activity incorporates oxygen into arachidonic acid that results in the formation of a labile intermediate peroxide, PGG2. The peroxidase activity reduces PGG2 to the endoperoxide product PGH2.39 This unstable compound is subject to further metabolism by at least nine terminal prostanoid synthases to form TX and the D, E, F, and I series PGs.24,49
While most human cells or tissues have the capacity to form an array of active prostanoids through tissue-specific expression of prostanoid synthases, the TX biosynthetic pathway predominates in platelets. The membrane-bound hemoprotein, thromboxane synthase (TXAS) that catalyzes the isomerization reaction of PGH2 to TXA2, also converts PGH2 to 12-l-hydroxy-5,8, 10-heptadecatrienoic acid (HHT) and malondialdehyde (MDA).50 However, the importance of HHT and MDA in the regulation of human platelet function is still poorly understood.
Despite its short half-life, PGH2 released from platelets can be also metabolized by PG synthases expressed outside of platelets in the plasma or in vascular cells. For example, platelets can contribute to the production of PGI2 in endothelial cells or in inflamed synovial cells in vitro through a transcellular shunt of PGH2.51,52 Platelet-derived PGH2 is also the substrate for PGD2 formation by plasma PGD synthase.21
PROSTANOID SIGNALING
Because of their short half-life, prostanoids do not circulate.53 They modulate cell function close to the site of their formation acting via specific G-protein-coupled receptors (GPCRs) on the membranes of their target cells (Table 34.1).24,49,54,55 A single gene
product receptor has been identified for TXA2 (the T prostanoid receptor or TP), prostacyclin (the IP), and PGF2α (the FP), while four distinct PGE2 receptors (the EP1-4) and two PGD2 (the DP1 and the DP2) have been cloned. Additional isoforms of the TP (α and β), FP (A and B), and EP3 (A-D) are generated through differential splicing.
product receptor has been identified for TXA2 (the T prostanoid receptor or TP), prostacyclin (the IP), and PGF2α (the FP), while four distinct PGE2 receptors (the EP1-4) and two PGD2 (the DP1 and the DP2) have been cloned. Additional isoforms of the TP (α and β), FP (A and B), and EP3 (A-D) are generated through differential splicing.
Table 34.1 G proteins associated with prostanoid receptors | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The sequences of the TPα and TPβ carboxy terminal splice variants diverge distal to the seventh transmembrane domain. Their expression is differentially regulated by two distinct promoters within the single human TP gene.56 Differences in signal transduction pathways have been noted,57 as have differential rates of agonist-induced desensitization.58 While both transcripts are present in human platelets, only the TPα protein has been detected.59 The TP couples to multiple G proteins to stimulate small G protein signaling pathways and may activate or inhibit adenylyl cyclase via Gs (TPα) or Gi (TPβ), respectively. In platelets, the TP interacts predominantly with Gq and G12/13. Gq activates protein kinase C-dependent pathways, which facilitate platelet aggregation, whereas G12/G13-mediated regulation of myosin light chain phosphorylation participates in receptorinduced platelet shape change. TXA2-induced platelet activation beyond the site of vascular injury is thought to be restrained by its short half-life (about 30 seconds) and by rapid TP desensitization. Heterodimerization with other GPCRs, such as the IP, may afford another mechanism to modulate downstream signaling.60 Additionally, endogenous inhibitors of platelet function, including PGI2 and nitric oxide, serve to limit platelet activation.20
Both PGI2 and PGE2 acting via the IP, or theoretically, via the DP1, the EP2, and/or the EP4 (all coupled via Gs to adenylate cyclase activation), may inhibit platelet function,24 while lower concentrations of PGE2 activate platelets via the EP3 in vitro.25 EP3 isoforms can couple via Gi or G12 to elevation of intracellular calcium, inhibition of cAMP generation, and activation of the small G protein Rho.
PROSTANOID DEGRADATION AND ELIMINATION
Thromboxane
TXA2 is rapidly hydrolyzed (T1/2 ˜30 seconds) to the stable and biologically inactive compound TXB2 in biologic fluids. Quantitation of TXB2 in serum or platelet-rich plasma permits assessment of the capacity of platelets to form TXA2 ex vivo.61,62,63,64 Typically, TXB2 formation is detected in serum derived from whole blood allowed to clot under standardized conditions or in platelet-rich plasma after induced platelet activation.63 These approaches have been used, for example, to validate the use of low doses of aspirin in cardiovascular prophylaxis65 or to reveal drug-drug interactions between low-dose aspirin and NSAIDs.66,67
Attempts to use TXB2 as an index of platelet activation in vivo, however, have proved unsuccessful. Platelets have the capacity to form TXA2 in concentrations of approximately 300 to 400 ng/mL blood,68 while maximal endogenous concentrations have been estimated to be three orders of magnitude lower, in the range of 1 to 2 pg/mL.69 This divergence between the capacity to generate TXA2 and the actual production rate in vivo is a feature of the feed forward, activation amplification role of TXA2 in the propagation of platelet-mediated clotting.70 Thus, the inevitable platelet activation that occurs during blood sampling, even under standardized conditions, results in TXB2 bursts that exceed circulating levels and render the measurement highly variable and essentially meaningless.71
The quantitation of urinary TXB2 metabolites, themselves not formed in whole blood, avoids this problem because TXB2 is rapidly cleared from the circulation.53,69 The first of two major pathways of TXB2 metabolism results in the formation of the β-oxidation products, 2,3-dinor TXB2, and 2,3,4,5-tetranor TXB2.72 The second involves the formation of molecules with a δ-lactone ring structure, of which 11-dehydro TXB2 is the major urinary product and the most abundant TXB2 metabolite in humans (2,3-dinor TXB2 is the most abundant metabolite in mice).72 Both, 2,3-dinor TXB2 and 11-dehydro TXB2 have been qualified as indices of TXA2 biosynthesis in vivo.69,71,73,74 Because neither metabolite is formed in the kidney, their detection in urine affords a noninvasive measure of systemic TXA2. 11-dehydro TXB2 itself undergoes extensive degradation, but its half-life in the circulation (˜60 minutes) exceeds that of 2,3-dinor TXB2 (15 to 17 minutes) and it is more abundant in urine.75,76 Thus, this metabolite is thought to be the more robust index of TXA2 formation in vivo.76
Experiments with low-dose aspirin revealed that roughly 80% of the systemic TX biomarker in urine derives from platelets under physiologic conditions.77 Excretion of urinary TX metabolites is augmented in clinical syndromes of platelet activation, including unstable angina and myocardial infarction,78 reperfusion following therapeutic thrombolysis,79 severe peripheral vascular disease,80 preeclampsia,81 pulmonary hypertension,82 sickle cell anemia,83 systemic lupus erythematosus,84 scleroderma,85 and stroke.86 Conversely, high levels of a urinary 11-dehydro TXB2, in patients on low-dose aspirin therapy have been linked to elevated cardiovascular risk.87
Prostacyclin
PGI2 undergoes a metabolic fate similar to that of TXB2. It is degraded hydrolytically to 6-keto-PGF1α and then enzymatically to 2,3-dinor-6-keto-PGF1α (6.8% of infused PGI2) and 15-ket o-13,14-dihydro-2,3-dinor-6-keto-PGF1α.53 While quantitation of 6-keto-PGF1α allows for monitoring the formation of PGI2 in vitro, urinary excretion of 2,3-dinor-6-keto-PGF1α has been qualified as an index of PGI2 formation in vivo.53 Quantitation of this metabolite revealed that the endogenous production of PGI2 under physiologic conditions is very low.53 The observation that its formation was markedly enhanced in clinical settings in which platelet-vascular interactions are increased, such
as unstable angina or myocardial infarction, suggested that PGI2 may act as an endogenous platelet-inhibitory agent, perhaps to restrain the propagation of a thrombotic response beyond the site of injury.88 Thus, concern was raised when clinical studies in healthy volunteers revealed unexpectedly that COX-2 selective NSAIDs depressed endogenous PGI2.89,90 These studies showed that COX-2 was the major source of PGI2 biosynthesis in humans, suggesting—before the first compound within this class was granted marketing approval—that COX-2 inhibition might elevate cardiovascular risk.18
as unstable angina or myocardial infarction, suggested that PGI2 may act as an endogenous platelet-inhibitory agent, perhaps to restrain the propagation of a thrombotic response beyond the site of injury.88 Thus, concern was raised when clinical studies in healthy volunteers revealed unexpectedly that COX-2 selective NSAIDs depressed endogenous PGI2.89,90 These studies showed that COX-2 was the major source of PGI2 biosynthesis in humans, suggesting—before the first compound within this class was granted marketing approval—that COX-2 inhibition might elevate cardiovascular risk.18
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


