Platelet Collagen Receptors
Stephen P. Watson
Richard W. Farndale
Masaaki Moroi
Stephanie M. Jung
Platelets, small anucleate circulating blood cells, are known to be activated by several physiologic substances, including ADP, thrombin, collagen, and adrenaline (epinephrine), as well as some nonphysiologic compounds. Collagen is thought to be one of the important physiologic activators because it powerfully induces platelet aggregation and is abundantly present in subendothelial tissue, which becomes exposed to the blood stream when blood vessels are damaged. Thus, exposed collagen fibers contribute to initial thrombus formation by interacting with circulating platelets. The mechanism of thrombus formation on collagen surfaces under blood flow has been studied by many investigators using ex vivo flow chamber systems designed to approximate the physiologic environment in blood vessels. This section provides a historical context for the platelet collagen receptors and subsequent sections provide details about our present knowledge of how these receptors initiate and facilitate the transmission of signals to result in thrombus formation.
After the high-affinity ligand-receptor interaction concept was established in the 1960s, such a stimulus-response reaction was also hypothesized for collagen-platelet interactions. Because platelets react specifically with collagen, a specific receptor for collagen was expected to be present on the platelet surface, but its identity remained elusive for many years because its ligand, collagen, a complex and insoluble macromolecule, is unlike classical ligands. This means that collagen may have numerous possible interactions, many of which would be of low affinity. Because of these obstacles, the collagen-platelet interaction was assessed only by collagen-induced platelet aggregation, not an ideal parameter since this response is influenced by numerous other platelet activating reactions, not only those directly related to platelet binding of collagen. Consequently, many proteins have been reported to be the candidates for platelet collagen receptors, even though they do not meet all the criteria for a physiologic receptor.
The first hypothesis, proposed by Jamieson et al.,1 suggested that the collagen receptor was an enzyme, a collagen glucosyltransferase that works as a receptor through an enzyme-substrate interaction. This hypothesis was supported by reports showing that a glycopeptide fragment of chicken collagen induced platelet aggregation just like native collagen fibers. Later, Chiang et al. purified a protein that specifically interacted with the chicken collagen fragment and proposed that this was the collagen receptor, but subsequent investigators could not verify that this was the case. During this period, other proteins were suggested to be receptors including, fibronectin, GPIIb/IIIa (integrin αIIbβ3), thrombospondin, factor XIII, propolypeptide of von Willebrand Factor (vWF), and CD36 (GPIV). Although these proteins interact with collagen, to date, none have been confirmed to contribute significantly to the platelet-collagen interaction, particularly under physiologic conditions. The major platelet collagen receptors were subsequently found by a combination of biochemistry and, in the case of glycoprotein VI (GPVI), careful studies of patient platelets deficient in the receptor, as discussed below.
INTEGRIN α2β1 (GPIa/IIa)
The integrins are heterodimers composed of an α and a β subunit selected from 18 α subunits and 8 β subunits, each a separate gene product, combining in 24 permitted pairings.2 Key species are α2β1, the only integrin collagen receptor expressed on the platelet; the fibronectin and laminin receptors, α5β1 and α6β1; and the fibrinogen receptor, αIIbβ3. α2β1 also has lamininbinding activity. The integrin subunits each have a short, conserved cytoplasmic domain, a single transmembrane span, and a long glycosylated stem supporting the head group, where the two subunits interact strongly and where ligand-binding occurs. The α subunit head is a 7-bladed β-propeller.
Integrins fall into two categories, defined by the presence within the α subunit of an I domain, so named because it is inserted between blades 2 and 3 of the β-propeller. This 200 amino-acid domain has a similar structure to the A domains of vWF, and is sometimes called the vWF A-domain. In these integrins, which include α2β1, the α subunit I domain contains the ligand-binding site,3 comprising an electronegative DxSxS motif that forms part of the Mg2+-binding metal iondependent adhesion site (MIDAS). This MIDAS Mg2+ ion is in turn coordinated by an amino-acid carboxylate anion in the ligand.
In the second category of integrins, those lacking an α subunit I domain, such as αIIbβ3 and α5β1, the ligand-binding site is found in a very similar structure, namely the β subunit I domain. For αIIbβ3, the ligand carboxylate anion might be the aspartate found within the RGD motif of the α chain of fibrinogen or in the C1 domain of vWF. Cooperative binding of such extended, polymeric proteins may occur through secondary ligand, for example, KQAGDV in the γ chain of fibrinogen4 binding to the α subunit β-propeller that forms the largest segment of the integrin head. Regulation involves the β subunit MIDAS, and two other cation-binding sites, the ADMIDAS (adjacent to MIDAS) and LIMBS (ligand-induced metal ion binding site).5
In 1985, Nieuwenhuis et al.6 reported a patient who had defective collagen-induced platelet aggregation and a bleeding tendency in association with a marked decrease in GPIa.*
Around the same time, Santoro7 provided biochemical evidence suggesting that GPIa interacts with collagen. Later, GPIa on the platelet membrane was shown to be present as a complex with GPIIa, the GPIa/IIa complex.8 Analyses of membrane proteins interacting with extracellular matrix proteins were performed for many different cells, leading to identification of very late activation antigens (VLA) of lymphocytes, cell adhesion receptors of leucocytes, and extracellular matrix receptors (ECMRs) of fibroblasts; the proteins interacting with collagen in these systems, VLA-2 and ECMR-II, were thus identified. It was subsequently discovered that GPIa/IIa, VLA-2, and ECMR-II were all the same protein, which was then designated as integrin α2β1, establishing that integrin α2β1 was a collagen receptor present in many cell types. The structural details of α2β1 are shown in FIGURE 30.1 and are described below, although one defining characteristic of α2β1, as related to its ability to interact with its ligand collagen, should be noted here. Since collagen is insoluble, the interaction of collagen and platelet α2β1 is usually analysed by adhesion assay, a condition in which platelet activation and interaction of α2β1 with collagen occur concomitantly and thus integrin activation cannot be differentiated from collagen adhesion. However, Stephanie Jung and Masaaki Moroi9 at Kurume University succeeded in separating these reactions by kinetic analyses with soluble collagen III monomers and demonstrated that α2β1 must be activated to exhibit high-affinity binding to collagen III, the resting state showing almost no binding activity toward this ligand.
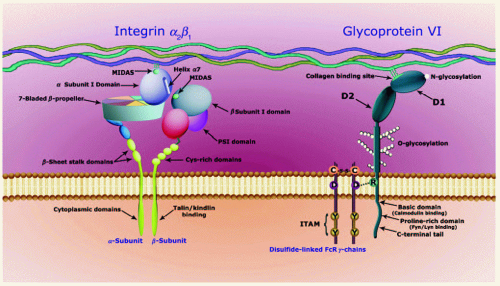 FIGURE 30.1 Structures of platelet collagen receptors. (Left) Integrin α2β1: the extracellular domain of the α2 subunit contains the I domain and its collagen-binding site. The I domain is located between blades 2 and 3 of the α subunit β-propeller. Helix 7, within the I domain, links the collagen-binding MIDAS with its counterpart in the β subunit I domain. The head of the β subunit also contains a hybrid domain (red), comprising polypeptide that is proximal to the I domain and a distal segment that leads into the plexin-semaphorin-integrin (PSI) domain, where the N-terminus is located. The β subunit stem contains cysteine-rich EGF domains. The cytoplasmic domains contain binding sites for numerous signalings, and adapter and cytoskeletal components, similar to focal adhesion components, and talin and kindlin binding regulate the activation of the integrin. (Right) Glycoprotein VI: The extracellular domain of GPVI contains two Ig-like domains (D1 and D2), and between the Ig-like domains and the transmembranes lies a mucin-like Ser/Thr-rich domain (O-glycosylation). A putative binding site for collagen is formed by interaction of two D1 domains when GPVI forms a dimer. The D1 domain may contain a glycosylation site (Asn72) for an N-linked carbohydrate chain. The presence of a charged residue (Arg252) in the transmembrane-membrane domain enables GPVI to associate with an FcR chain, which contains a negatively charged Asp residue. The cytoplasmic portion of GPVI consists of four regions: juxtamembrane portion; basic region, which contains a calmodulin binding site; proline-rich domain, to which Fyn and Lyn bind to through their SH3 domains; and the C-terminal region. Mouse GPVI is truncated, lacking the latter domain. |
GLYCOPROTEIN VI
Although α2β1 was established as a collagen receptor on platelets, there was a report suggesting the presence of an approximately 60-kDa collagen-reactive platelet protein on platelets based on patient studies. In 1987, Sugiyama et al.10 reported a patient
with idiopathic thrombocytopenic purpura (ITP) whose platelets were defective in collagen-induced aggregation. Her serum contained an antibody to a platelet protein of approximately 60 kDa, although her own platelets did not contain this antigen and lacked reactivity to collagen. These results suggested that the antigen, designated P62, may be a receptor for collagen or a protein involved in a collagen-platelet interaction. This was supported by studies of another patient whose platelets were deficient in GPVI. No one had studied the characteristics of this protein at that time, but it was named according to its mobility in nonreduced-reduced two-dimensional SDS gel electrophoresis according to the convention of Phillips and Agin.11 The second patient’s platelets also lacked collagen-induced aggregation, while showing no abnormalities in aggregation induced by other platelet agonists. The antibody from the serum of the first patient10 did not react with platelets from the GPVI-deficient patient, indicating that P62 and GPVI were the same proteins. These results strongly suggested that GPVI was a newly defined platelet collagen receptor.
with idiopathic thrombocytopenic purpura (ITP) whose platelets were defective in collagen-induced aggregation. Her serum contained an antibody to a platelet protein of approximately 60 kDa, although her own platelets did not contain this antigen and lacked reactivity to collagen. These results suggested that the antigen, designated P62, may be a receptor for collagen or a protein involved in a collagen-platelet interaction. This was supported by studies of another patient whose platelets were deficient in GPVI. No one had studied the characteristics of this protein at that time, but it was named according to its mobility in nonreduced-reduced two-dimensional SDS gel electrophoresis according to the convention of Phillips and Agin.11 The second patient’s platelets also lacked collagen-induced aggregation, while showing no abnormalities in aggregation induced by other platelet agonists. The antibody from the serum of the first patient10 did not react with platelets from the GPVI-deficient patient, indicating that P62 and GPVI were the same proteins. These results strongly suggested that GPVI was a newly defined platelet collagen receptor.
There were essentially no studies on GPVI for some years after its discovery because of the lack of tools to analyze this protein except for the patient autoantibody. While searching for platelet-reactive sites of collagen, Michael Barnes and Richard Farndale at Cambridge found that a collagen-mimetic peptide that was called collagen-related peptide (CRP), containing 10 repeats of the sequence Gly-Pro-Hyp (GPO), induced platelet aggregation and this reaction was independent of α2β1.12 Finally, the era of intense investigations on GPVI as platelet collagen receptor was initiated. Steve Watson’s group in Oxford identified the Fc receptor γ chain as a critical component in collagen signaling in platelets that shortly thereafter was shown to associate with GPVI and to be essential for platelet activation.13,14,15 CRP reacted specifically with GPVI16 and could stimulate tyrosine phosphorylation in platelets in a manner similar to collagen.17 A snake venom protein, convulxin, first identified in the late 1970s, was subsequently found to induce a pattern of tyrosine phosphorylation similar to that induced by collagen, including phosphorylation of the FcR γ chain. The identification of materials that specifically reacted with GPVI and activated platelets facilitated the purification and cloning of GPVI18 and also the analysis of its function.
Human GPVI is a member of the immunoglobulin superfamily and is encoded in the leucocyte receptor complex on chromosome 19; the FcR γ chain is encoded on chromosome 1. GPVI possesses an N-terminal and a C-terminal extracellular Ig domain, designated D1 and D2, linked by a single peptide strand that allows some flexibility in the head of the receptor (FIGURE 30.1). A glycosylated stem connects D2 to the transmembrane domain and thence to the relatively short (50 amino acids) cytoplasmic domain that can bind calmodulin close to the plasma membrane, and the Src family kinases Fyn and Lyn through a proline-rich domain closer to the C-terminal cytoplasmic tail (see below). The transmembrane domain contains an arginine residue that forms a salt bridge with an aspartate residue in the equivalent location of FcR γ chain and supports the association between the two.18 Coexpression of FcRγ with GPVI is required for cell-surface expression of the latter in platelets.
In 2006, the crystal structure of GPVI was described by Andrew Herr et al.19 revealing interactions between two copies of the receptor that might form the basis for physiologic dimerization.20 A collagen binding site, located largely within D1, was modeled onto the receptor, consistent with the effect of site-directed mutagenesis on ligand binding.21 Whether this model properly reflects the interaction of GPVI with insoluble collagen has yet to be resolved. More recently, a crystal structure and binding study of LAIR1, a single Ig domain receptor that shares substantial homology with D1 of GPVI, led to the suggestion of a nearby but discrete ligand-binding site in GPVI.22 These differing proposals will be fruitful areas for research.
GPVI has also been suggested to bind laminin, which is a major component of the basement membrane.23 The interaction with laminin is dependent on integrin α6β1 and leads to platelet spreading, but laminin is unable to induce aggregation of platelets in suspension, possibly because it has a 10-fold lower affinity for GPVI than collagen. Laminin is the major platelet-reactive component in the basal lamina, and, like collagen IV, is unable to activate platelets (see below). The physiologic significance of the interaction of platelets with laminin may be to mediate sealing and repair of small areas of damage to the vessel in the absence of thrombus formation.
Thus, platelets have two collagen receptors: α2β1 that has higher affinity for collagen, but requires activation for its affinity to be fully expressed, and GPVI that activates platelets. Kinetic studies using recombinant GPVI indicated that the immunoglobulin receptor had a weaker affinity for collagen than α2β1.24 Under physiologic conditions, these two receptors would work synergistically, with α2β1 supporting adhesion and GPVI supporting activation, and thus each contributing to effective thrombus formation. The use of GPVI- and α2β1-specific peptides as surface coatings for in vitro flow studies reveals the need for both receptors along with vWF for full platelet aggregation to take place.25
There is also evidence in the literature that platelets may express one or more further receptors for collagen that may support adhesion and possibly activation. For the most part, this evidence is linked to a single study or a handful of studies that have implicated certain proteins, such as GPV, or have demonstrated signaling events that cannot be attributed to activation of GPVI and integrin α2β1.26 The overall significance of these putative receptors in platelet activation by collagen is uncertain and has been reviewed elsewhere.27
THE CLASSIC TWO-STEP, TWO-SITE MODEL AND THE CURRENT MODEL FOR PLATELET ADHESION
Any model proposed for how the platelet collagen receptors induce platelet activation must take into account the differential roles of α2β1 and GPVI. In 1989, Michael Barnes reported that platelets could adhere to several specific cyanogen bromide (CB) peptides of collagen I and III, but only a subset of these peptides exhibited aggregatory activity.28 This suggested that the collagen molecule may contain sites specific for platelet adhesion that are different from those that stimulate aggregation. Around this time, Samuel Santoro proposed the two-step, two-site model, based on chemical modification of collagen,29 in which collagen has a higher-affinity binding site for α2β1, for the adhesion reaction, and a lower-affinity binding site for a second receptor, presumably the one responsible for platelet activation. The development of peptide libraries (Toolkits)30 based on the primary sequence of collagen type III by Richard Farndale’s
group at Cambridge further advanced our understanding of the initial binding interaction between platelets and collagen. The various α2β1-binding sites were defined,31,32 and the activating site within CB4 of collagen III was identified as binding GPVI.33 After GPVI was found to be a collagen receptor on platelets, this two-step, two-site model was applied to GPVI and integrin α2β1 to explain their separate roles in the platelet-collagen interaction: α2β1 would bind first, assisting the other collagen receptor GPVI to bind and initiate the activation of platelets. However, the finding that integrin α2β1 exists in a resting state that exhibits low affinity for collagen III and an activated high-affinity state induced by platelet agonists mandated a reappraisal of this model: it is now thought that platelets first interact with collagen through GPVI, activate α2β1, and the activated α2β1 binds strongly to collagen, resulting in initiation of a cascade of reactions to activate platelets. However, it should be noted that the traditional two-step, two-site model and the currently accepted model are not mutually exclusive, since it may be possible that both types of receptor-collagen interactions may occur under physiologic conditions. Even in nominally “resting” platelets, the presence of a low level of the active, high-affinity form of α2β1 may contribute to platelet adhesion to collagen and it is also possible that the resting receptor may be able to interact with the higher-affinity binding sequences in collagen type I (see below).
group at Cambridge further advanced our understanding of the initial binding interaction between platelets and collagen. The various α2β1-binding sites were defined,31,32 and the activating site within CB4 of collagen III was identified as binding GPVI.33 After GPVI was found to be a collagen receptor on platelets, this two-step, two-site model was applied to GPVI and integrin α2β1 to explain their separate roles in the platelet-collagen interaction: α2β1 would bind first, assisting the other collagen receptor GPVI to bind and initiate the activation of platelets. However, the finding that integrin α2β1 exists in a resting state that exhibits low affinity for collagen III and an activated high-affinity state induced by platelet agonists mandated a reappraisal of this model: it is now thought that platelets first interact with collagen through GPVI, activate α2β1, and the activated α2β1 binds strongly to collagen, resulting in initiation of a cascade of reactions to activate platelets. However, it should be noted that the traditional two-step, two-site model and the currently accepted model are not mutually exclusive, since it may be possible that both types of receptor-collagen interactions may occur under physiologic conditions. Even in nominally “resting” platelets, the presence of a low level of the active, high-affinity form of α2β1 may contribute to platelet adhesion to collagen and it is also possible that the resting receptor may be able to interact with the higher-affinity binding sequences in collagen type I (see below).
THE ROLE OF COLLAGEN IN THROMBUS FORMATION
The molecular events underlying thrombus formation in the high flow rates in arteries and arterioles are depicted in FIGURE 30.2. Platelets marginate along the vessel walls because of the large mass of red blood cells that flows in the centre of the vessels. Platelets are kept in a quiescent state by the continuous release of nitric oxide and prostacyclin from the vascular endothelial cells and by removal of low levels of ADP and ATP that have been released from damaged cells by apyrase-like activity of endothelial CD39. Damage to the endothelial surface exposes collagen fibers that capture vWF from the flowing blood through its collagen-binding A3 domain.
 FIGURE 30.2 Role of collagen in platelet activation. Damage to the endothelial cell wall exposes subendothelial collagen fibers that bind to von Willebrand factor (vWF) in the bloodstream. Platelets are captured (or tethered) through the interaction of vWF and GPIb-IX-V complex on the platelet surface. This enables binding of collagen to GPVI, which stimulates activation of integrins αIIbβ3 and α2β1. In turn, the integrins bind to vWF and collagen, respectively, resulting in a stable adhesion. GPVI also promotes platelet spreading and secretion of the feedback agonists ADP and TxA2 that activate circulating platelets. Cross-linking of platelets by fibrinogen mediates platelet aggregation. |
vWF mediates platelet tethering through a very fast on-rate of association to the GPIb-IX-V complex on the platelet surface. The GPIb-IX-V complex is a unique receptor in the genome and mutations in GPIbα, GPIβ, or GPIX subunits give rise to the severe bleeding disorder Bernard Soulier syndrome due to loss of the capacity for platelet tethering, as well as variable thrombocytopenia. The interaction between GPIbα and the A1 domain of vWF, however, does not lead to rapid, stable adhesion because of a fast off-rate of dissociation. This can be illustrated experimentally by the rolling of platelets under flow conditions on an immobilized vWF surface.
In the subendothelial wall, platelet tethering facilitates the interaction of fibrillar collagen with the low-affinity GPVI receptor complex, stimulating powerful activation of the integrins α2β1 and αIIbβ3. These two integrins mediate stable adhesion through binding to collagen and vWF, respectively. Additionally, GPVI stimulates platelet spreading, thereby strengthening the interaction with the subendothelial matrix. The action of GPVI is supported by much weaker signals from the GPIb-IX-V complex and integrins α2β1 and αIIbβ3. The relative contribution of each receptor to platelet activation is unclear as all four receptors signal through overlapping pathways with critical roles for Src and Syk tyrosine kinases, and phospholipase Cγ2 (PLCγ2).
Platelet activation by GPVI causes ADP release from platelet dense granules and de novo synthesis of TxA2 from arachidonic acid. These secondary mediators interact synergistically to mediate activation of platelets that have become newly tethered to αIIbβ3-bound vWF. In turn, plasma fibrinogen, which is present in molar excess over other adhesion proteins, mediates platelet aggregation by cross-linking the integrin αIIbβ3. Thus, aggregate formation is critically dependent on the secondary mediators ADP and TxA2 that signal through the P2Y1 and P2Y12 receptors and the TP thromboxane receptors, respectively. P2Y1 and TP receptors signal through activation of PLCγ isoforms downstream of the heterotrimeric G protein, Gq. The P2Y12 receptor signals through the Gi family of heterotrimeric G proteins and undergoes a powerful synergy with signals from both Gq and tyrosine kinase-linked receptors.
Activated platelets provide a procoagulant surface that leads to formation of thrombin through the coagulation cascade. Thrombin reinforces platelet activation through the protease activated receptors (PARs), PAR-1 and PAR-4, which are also coupled to Gq. In addition, thrombin cleaves fibrinogen to fibrin, thereby further strengthening the growing thrombus, and this is further cemented by clot retraction mediated through actin-myosin contraction mediated downstream of integrin αIIbβ3. Together these interactions generate a robust thrombus that is able to withstand the high shear forces found within the arteriolar circulation.
In situations of mild injury, the roles of GPVI and integrin α2β1 are redundant with those of other signaling receptors and adhesive integrins, respectively. The level of expression of α2β1 on the platelet surface is approximately 1% to 2% of that of integrin αIIbβ3, which therefore dwarfs its role in supporting adhesion. This is illustrated by the absence of patients
with platelet-bleeding disorders that can be attributed solely to a reduction in functional α2β1, whereas there are several hundreds of patients with function disrupting mutations in integrin αIIbβ3, a condition known as Glanzmann thrombasthenia. Very few patients have also been reported with functional defects in GPVI associated with a bleeding diathesis. Furthermore, in the majority of these cases, the loss of GPVI is mediated by an immune thrombocytopenia and is associated with a marked reduction in platelet count. There are currently just two patients with a bleeding diathesis in which the only known platelet defect is the loss of functional GPVI as a result of inheritance of function-disrupting mutations in the glycoprotein receptor. This very low number of patients can be explained by redundancy with other platelet signaling receptors such as those for thrombin, which is generated by exposure of tissue factor on the surface of subendothelial smooth muscle and other cells, and therefore also plays a role in the initial events that trigger activation.
with platelet-bleeding disorders that can be attributed solely to a reduction in functional α2β1, whereas there are several hundreds of patients with function disrupting mutations in integrin αIIbβ3, a condition known as Glanzmann thrombasthenia. Very few patients have also been reported with functional defects in GPVI associated with a bleeding diathesis. Furthermore, in the majority of these cases, the loss of GPVI is mediated by an immune thrombocytopenia and is associated with a marked reduction in platelet count. There are currently just two patients with a bleeding diathesis in which the only known platelet defect is the loss of functional GPVI as a result of inheritance of function-disrupting mutations in the glycoprotein receptor. This very low number of patients can be explained by redundancy with other platelet signaling receptors such as those for thrombin, which is generated by exposure of tissue factor on the surface of subendothelial smooth muscle and other cells, and therefore also plays a role in the initial events that trigger activation.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


