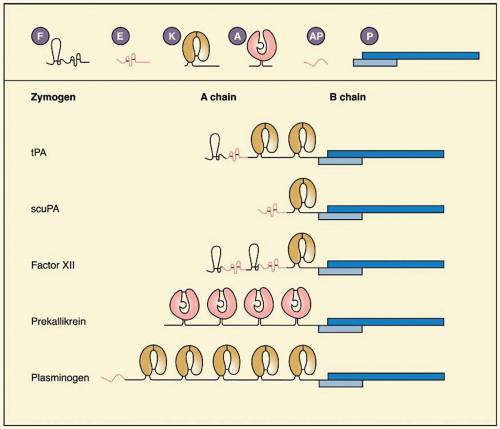
 FIGURE 20.1 Structural elements of several fibrinolytic serine proteases. A, apple domain (˜80 residues); AP, activation peptide; E, epidermal growth factor domain (˜32 residues); F, fibronectin finger domain (˜40 residues); K, kringle (˜90 residues); P, protease domain (B chain; ˜250 residues); scuPA, single-chain urokinase; tPA, tissue-type plasminogen activator. |
in more detail below). The affinity of the kringles for lysine is exploited in affinity chromatography.29 The kringles also bind to ω-aminocarboxylic acids, such as 6-aminohexanoic acid (εACA, 6-AHA), p-aminomethyl benzoic acid (p-AMBA), and trans-4-amino-methyl-cyclohexane-carboxylic acid (trans-AMCA, also known as tranexamic acid). These are used experimentally and clinically to inhibit the functional activity of plasmin by competing with the binding of plasmin(ogen) to fibrin and cell surfaces.
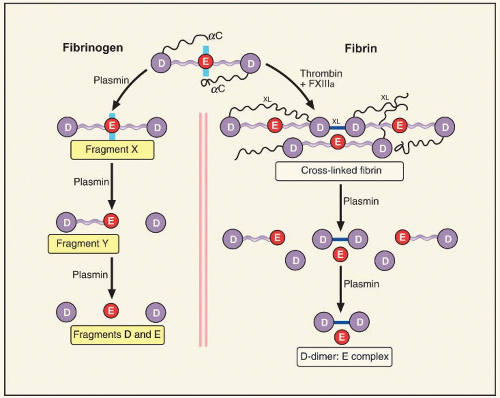 FIGURE 20.2 Degradation of fibrin(ogen) by plasmin. The principal structure of fibrinogen (top) is a three domain globular protein with extending αC domains. Through the action of plasmin the degradation fragments D, E, α-helical coiled coils that connect them, and the Aα chains that extend from the carboxy-terminus of the D-domains. Fibrinogen is degraded asymmetrically (left panel). Intermediate degradation product, fragment X, consists of all three domains connected by coiled coils, but lacks the Aα chains and the Bβ1-42 sequence. Fragment Y is composed of the central E-domain connected by a coiled coil to the D-domain. Cross-linked fibrin (right panel), occurs through the enzymatic actions of thrombin and activated factor XIII (FXIIIa). Thrombin cleaves fibrinopeptides A and B from the E-domain, and FXIIIa cross-links (XL) fibrin longitudinally between D-domains and within the α-chain extensions (see Chapter 16). Plasmic cleavage of the two-stranded protofibrils initially removes the cross-linked α chains, then the coiled coils to liberate a series of FDPs, the smallest of which is DD/E. Larger complexes, such as DY/YD, are also liberated from cross-linked fibrin, but these are subsequently degraded to the DD/E moiety. |
Activation and Regulation of Fibrinolysis” At this stage of our discussion, it can be assumed that the binding is primarily dependent on exposed lysine residues, and that the binding to fibrin and to cell receptors for PLG are generally equivalent.
Table 20.1 Properties of proteins of the fibrinolytic system | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
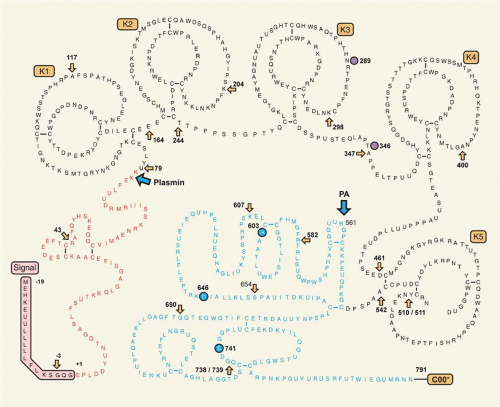 FIGURE 20.3 Sequence of the human PLG molecule. Positions of the 18 introns are indicated by yellow arrows at (for type 1 and 2 introns) and between (for type 0 introns) residues. The 19-residue signal peptide (shown in the pink box with negative numbers) is cleaved by the signal peptidase at the Gly-Glu peptide bond. Conversion of PLG to plasmin by PAs occurs after the peptide bond Arg561-Val562 (shown by a blue arrow). Plasmin cleaves primarily at Lys77-Lys78 bond (shown by a blue arrow) to release the activation peptide (red lettering) generating Lys-plasmin(ogen). K1-K5 kringles are located in the A chain (black lettering). The B chain remains attached to the A chain (blue lettering) after cleavage at 561-562 by PAs via the disulphide bonds Cys548-Cys666 and Cys558-Cys566. Carbohydrate attachment sites (Asn289 and Thr346) are shown as purple circles. The residues that comprise the classical serine protease Ser-His-Asp catalytic triad are circled in blue. (Redrawn based on Petersen TE, Martzen MR, Ichinose A, et al. Characterization of the gene for human plasminogen, a key proenzyme in the fibrinolytic system. J Biol Chem 1990;265:6104.) |
vessel surface.55,56,57 Additional glycosylation sites at Ser24958 and Ser33959 have been described; a further serine residue, Ser578, can be phosphorylated.60
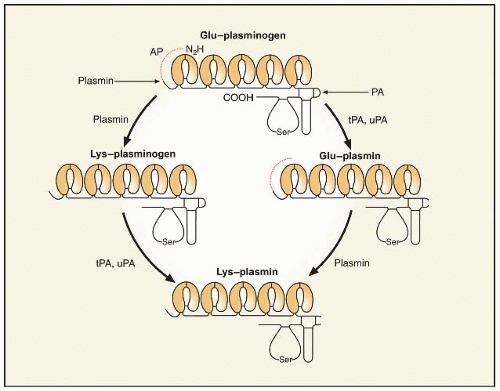 FIGURE 20.4 Activation of native Glu-PLG. PA cleaves at Arg561-Val562, separating the B (light, protease, or catalytic) and the A (heavy, kringle) chains. Glu-PLG and Glu-plasmin forms both contain the amino-terminal activation peptide from Gln1 to Lys76 (shown in red). Plasmin has the potential to cleave this activation peptide (left side), producing Lys-PLG, an intermediate form that interacts with fibrin more efficiently and is readily cleaved by the PAs (tPA and uPA). Plasmin can also cleave the activation peptide from Glu-plasmin, generating Lys-plasmin (right side). The five kringle structures of the A chain are involved in binding of PLG to both fibrin and cell receptors. The catalytic center contains the typical Ser-His-Asp residues and is the major site of interaction with α2antiplasmin. |
XII (FXII), prekallikrein (PK), and high molecular weight kininogen (HK), is known as the contact PA pathway. There are also PAs from bacteria and the vampire bat, which are briefly included here. The two human proteases, tPA and uPA, are also implicated respectively in neurobiology (for review see Ref. 90) and tumor biology (for review see Refs. 91 and 92). These aspects are not considered further in this chapter, which focuses on PAs in hemostasis.
Table 20.2 Congenital PLG mutations | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
concentrations, and nature of the fibrin stimulator. In the absence of fibrin, KM values for the activation of Glu-PLG by tPA range from 9 µM to slightly more than 100 µM.125,157,158 In general, KM is three to four times lower with tPA than with sctPA for the activation of Glu-PLG in the absence of fibrin. In the presence of fibrin, this difference is less apparent with KM values typically two orders of magnitude smaller, with only moderate increases of kcat. Values for KM in the presence of fibrin range from 0.16 to 1.1 µM PLG, and kcat values from 0.1 to 1.1 per second.125,158 Several authors found nonlinear enzyme kinetics,156,157,159 and there appear to be two phases in the activation of Glu-PLG by tPA in the presence of fibrin, with an initial KM of 1.05 µM and kcat of 0.15 per second. Later in the process, as new high-affinity binding sites for PLG and tPA are exposed in partially digested fibrin,160,161,162,163,164 KM decreases to 0.07 µM, whereas the kcat is unchanged.159 The key message is that PLG is not readily activated by tPA except in the presence of fibrin, because it is only in this situation that the KM for the reaction is consistent with the circulating PLG concentration of 2 µM.
activity. Lys300, situated in the flexible loop region 297 to 313, forms a weak interaction with Asp355 and pulls the adjacent active site Ser356 into the position found in the fully active protease. Site-directed mutagenesis of Lys300 to Ala201 or of Asp355 to Asn202 resulted in a 40-fold and 270-fold reduction in activity, respectively, compared to wild-type scuPA. Altering the flexibility of the 297-313 loop changes the intrinsic catalytic activity of scuPA.203
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


