INTRODUCTION
SUMMARY
Plasma cell neoplasms are tumors derived from an expansion of mutated mature B-cells and their precursors. These neoplasms include essential monoclonal gammopathy (synonym: monoclonal gammopathy of unknown significance; Chap. 106), smoldering myeloma (Chap. 107), myeloma (Chap. 107), solitary and extramedullary plasmacytomas (Chap. 107), light-chain amyloidosis (Chap. 108), and Waldenström macroglobulinemia (Chap. 109). The prototype of a malignant plasma cell neoplasm is myeloma, which is characterized by complex genetic alterations, best assessed by metaphase cytogenetics, fluorescence in situ hybridization analysis, and gene-expression profiling. The genetic changes are more akin to solid tumors than to hematologic malignancies. Interactions between myeloma cells and the marrow microenvironment affect the survival, proliferation, and drug resistance of myeloma cells, and the development of osteoporosis or osteolysis, which is a hallmark of myeloma. As in most malignancies, a cancer stem cell (e.g., myeloma stem cell) has been identified and is the most likely site of drug resistance, which almost invariably develops during treatment; such cells are not affected by the typical drugs one uses in patients with myeloma. The best prognostic markers in myeloma in order of importance are the presence of (1) specific cytogenetic abnormalities, (2) extent of the disease by appropriate imaging techniques, such as magnetic resonance imaging and/or combined positron emission and computed tomographic imaging, (3) the serum free light-chain level and kappa-to-lambda ratio, and (4) the use of the International Staging System. The development of several classes of drugs over the past decade in combination with transplantation, has improved therapeutic outcomes significantly in patients achieving an unequivocal complete remission. Thus, optimal techniques to assess minimal residual disease have also become important.
Acronyms and Abbreviations
AL, light-chain amyloidosis; BAFF, B-cell activating factor; BCR, B-cell receptor; BMSC, bone mesenchymal stem cell; BTK, Bruton tyrosine kinase; CDR, complementarity determining regions of the heavy chain; CR, complete remission; CSC, cancer stem cell; D, diversity immunoglobulin gene segment; FISH, fluorescence in situ hybridization; FLC, free light chain; GFR, glomerular filtration rate; ICAM-1, intercellular adhesion molecule 1; Ig, immunoglobulin; IGH, immunoglobulin heavy chain; IGF-1, insulin-like growth factor 1; IL, interleukin; IRAK, interleukin-1 receptor-associated kinase; JAK2/STAT3, Janus kinase 2/signal transducers and activators of transcription; JH, joining region immunoglobulin gene segment; M, monoclonal; MBD, myeloma bone disease; MPC, multiparameter flow cytometry; MRD, minimal residual disease; MRI, magnetic resonance imaging; mSMART, Mayo stratification of myeloma and risk-adapted therapy; MYD, myeloid differentiation primary response gene; nCR, near complete remission; NEK2, a serine/threonine kinase; NF-κB, nuclear factor κB; OB, osteoblast; OC, osteoclast; OL, osteolytic lesion; OPG, osteoprotegerin; PCN, plasma cell neoplasm; PDGF, platelet-derived growth factor; PET/CT, 18F-fluorodeoxyglucose positron emission tomography–computed tomography; pP-7, a hyperphosphorylated protein; RAG, recombinase-activating genes; RANK, receptor activator of NF-κB; RARα, retinoic receptor α; RB, retinoblastoma gene sCR, stringent complete remission; sIFE, serum immunofixation electrophoresis; SMM, smoldering myeloma; SP, side population; SPEP, serum protein electrophoresis; TGF-β, transforming growth factor β; TLR, toll-like receptor; TME, tumor microenvironment; TNF-α, tumor necrosis factor α; TRAF3, the adaptor molecule for toll receptor; uIFE, urine immunofixation electrophoresis; UPEP, urine protein electrophoresis; VCAM-1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor; VH, the variable immunoglobulin gene segment.
DEFINITION AND HISTORY
Plasma cell neoplasms (PCNs) are clonal B-cell tumors that range from stable disease without functional abnormalities (monoclonal gammopathy, synonym monoclonal gammopathy of unknown significance) to one of slowly proliferating plasma cells (smoldering myeloma [SMM]), to one resulting in end-organ compromise (myeloma). PCNs are accompanied by the synthesis and release into the plasma of a monoclonal (M) protein, and, in the case of myeloma, either diffuse osteoporosis or osteolytic lesions. Myeloma accounts for approximately 1 percent of all malignant diseases and 10 percent of hematologic malignancies.
Approximately two-thirds of patients presenting with an M protein have (1) monoclonal gammopathy (Chap. 106), whereas approximately 15 percent have (2) myeloma (Chap. 107). Other diseases associated with M-protein productions are (3) immunoglobulin light-chain amyloidosis (AL) (10 percent; Chap. 108) resulting from the deposition of immunoglobulin (Ig) fragments in visceral organs a consequence of extensive misfolding of these Ig fragments, (4) SMM (3 percent; Chap. 107), (5) Waldenström macroglobulinemia (3 percent; Chap. 109), (6) lymphoproliferative disorders (2 percent; Chap. 90), (7) solitary or extramedullary plasmacytomas (1 percent; Chap. 107) and (8) miscellaneous other diseases (2 percent).3
NORMAL B-CELL DEVELOPMENT
B-cell development is discussed in detail in Chaps. 74 and 75. In brief, B-cell lymphopoiesis occurs initially in the marrow and in lymphoid tissues. In the marrow, the pro–B-cell, undergoes rearrangement of immunoglobulin heavy chain (IGH) genes and, then, is designated a pre–B-cell, which is characterized by the presence of cytoplasmic μ chains. Subsequent rearrangement of the light chain enables the cell to express surface IgM, the immature B lymphocyte phase of development. These cells leave the marrow and upon entering the blood express surface IgD, which then defines them as virgin B cells, also characterized by G0 cell-cycle arrest. Virgin B cells enter the lymphoid tissue, where they are exposed to antigen-presenting cells, become activated when in contact with the corresponding antigen and differentiate into short-lived, low-affinity plasma cells or memory B-cells. These memory B-cells travel from the extra-follicular area of the lymph node to the primary follicles, where if confronted with an antigen, presented by follicular dendritic cells, a secondary response is induced. At this stage, primary follicles change into secondary follicles containing germinal centers. Through activation by an antigen, the memory B cells differentiate into centroblasts, resulting in Ig isotype switching and somatic mutations in the variable region of the immunoglobulin gene with the generation of high-affinity antibodies. Centroblasts progress to the centrocyte stage and reexpress surface Ig. The centrocytes with high-affinity antibodies differentiate into either memory B cells or plasmablasts, which then move to the marrow and terminally differentiate to plasma cells. Marrow plasma cells produce most of the plasma immunoglobulins and have a life span of approximately 3 weeks.
Three distinct gene segments, the variable (VH), diversity (D), and joining region (JH) genes, encode the variable region of the heavy chain, whereas two segments, variable (Vκ or V) and joining (Jκ or J) region genes, encode the variable fraction of the light chain. The IGH locus on chromosome 14q32 contains an estimated 100 to 150 VH genes, 30 D, and six JH gene segments. Because some of the VH genes are nearly identical, it is likely that 60 to 70 VH genes are available for rearrangement. These 60 to 70 genes belong to seven families (VH 1 to 7) whose members have more than 80 percent sequence homology. Of the 75 known Vκ sequences, only 36 are potentially functional and of the 36 known V sequences, only 24 are functional. Rearrangement of the V gene segments is dependent on the protein products of the recombinase-activating genes RAG-1 and RAG-2. Recombination of V genes starts in lymphoid progenitors within the IGH locus of either the maternal or paternal chromosome 14. If the initial VH-D-JH rearrangement yields a sequence that cannot be translated, then rearrangement of the IGH locus proceeds on the other allele. The presence on the B-cell surface of a fully assembled μ heavy chain rearrangement begins when one of the Vκ genes rearranges to one of the Jκ genes. If κ light-chain rearrangement is unsuccessful on both alleles, by default light chains will subsequently rearrange. The Ig heavy and light chains each contain three hypervariable complementarity determining regions segments, which are the areas of the immunoglobulin in direct contact with the antigen. In a process of trial and error, immunoglobulins increase their affinity for an antigen by a series of somatic mutations. The complementarity determining regions of the IGH (CDR) 3 is the most variable portion of the Ig molecule, because it not only contains somatic mutations as is the case for CDR 1 and CDR 2, but it also encompasses the 3′ end of VH, all of D, and the 5′ end of JH. It is, therefore, an ideal marker to detect a very small population of the malignant myeloma clone within a larger population of normal cells.
ETIOLOGY AND PATHOGENESIS
Although monoclonal gammopathy (Chap. 106) shares the same constellation of risk factors and cytogenetic abnormalities with myeloma, it is an antecedent neoplasm that may undergo clonal evolution to any one of the PCNs or to a B-cell lymphoma.1 Two studies have reported that monoclonal gammopathy is a precursor to myeloma in virtually all cases.23
Retrospective population-based cohort studies have established that nearly 80 percent of cases of myeloma develop from IGH monoclonal gammopathy. The remaining 20 percent have serum free light chain (FLC) monoclonal gammopathy. The prevalence of FLC monoclonal gammopathy is 0.8 percent in the general population. It progresses to myeloma in a minority of patients at the rate of 0.3 percent per year,3 much lower than the conventional monoclonal gammopathy progression rate of approximately 1 percent.
Factors such as chronic antigen stimulation and chemical exposure have been suspected in the development of monoclonal gammopathy and other PCNs. Some studies have found a positive association,45 but the results have not been consistent and given our current understanding of the genetic precedents of myeloma, one would have to show a direct effect of such agents on the causal mutations involved.
A familial history of monoclonal gammopathy and myeloma has been reported to be a risk factor for developing the disease in first-degree relatives, including a population-based study from the Mayo Clinic. A twofold increased relative risk was noted for the development of monoclonal gammopathy among the first-degree relatives of myeloma and monoclonal gammopathy patients. In a large Swedish population study, among first-degree relatives of patients with monoclonal gammopathy, a threefold increased risk for both monoclonal gammopathy and myeloma, a fourfold risk of developing Waldenström macroglobulinemia, and a twofold risk of developing B-cell chronic lymphocytic leukemia was observed.6 These observations support the role of both germline susceptibility genes and possibly immune-related phenomena in the causation.7 Because of the extremely low lifetime-risk of developing myeloma in the general population (0.2 percent), it would be inefficient to screen the first-degree relatives of persons with myeloma or monoclonal gammopathy.
Hyperphosphorylated paratarg-7, a frequent autoantigenic target of human paraproteins, is linked to both familial and nonfamilial forms of monoclonal gammopathy and myeloma.7 Paratarg-7 is the target of 15 percent of the monoclonal proteins of the IgA and IgG type, and 11 percent of IgM-monoclonal gammopathy or macroglobulinemia patients. All patients with paratarg-7–specific paraproteins were carriers of a hyperphosphorylated protein (pP-7); this hyperphosphorylation is inherited in a dominant fashion. Hyperphosphorylation is a result of inactivation of phosphatase 2A.8 Hyperphosphorylated paratarg-7 carriers are most prevalent among Americans of African wdefined single risk factor for monoclonal gammopathy or myeloma in all ethnic groups9 and is associated with a sixfold increased risk of IgM monoclonal gammopathy or macroglobulinemia.10
Nearly 50 percent of patients with monoclonal gammopathy have plasma cells with translocations involving the IGH locus on chromosome 14q32 and one of the five partner chromosomes: 11q13 (cyclin D1 gene), 4q16,3 (FGFR-2 and MMSET), 6q21 (CCND3), 16q23 (c-maf), and 20q11 (maf-B).11,12,13,14
Unfortunately, none of the molecular or chromosomal abnormalities associated with myeloma predict the evolution of monoclonal gammopathy to myeloma. Two clinical risk stratification models propose high-risk features that can predict progression from monoclonal gammopathy and SMM to myeloma.15,16 While one model uses the type of immunoglobulin, quantity of M-protein, and the serum FLC ratio to determine the risk of progression, the other model is based on flow cytometry findings of aberrant plasma cells, marrow plasma cell percentage, DNA aneuploidy, and immune paresis (a decrease in noninvolved immunoglobulins).
SMM is discussed in Chap. 107. In addition to the marrow plasma cell burden and quantitative M-protein (>3 g/dL), presence of light-chain proteinuria and IgA M-heavy chain were identified as separate risk factors predicting progression to active myeloma.17,18,19,20 The median time of progression to myeloma has been reported to range between 3 and 8 years in low-risk groups, and between 1 and 2 years in high-risk groups.17,18,19,20,21,22 Some studies have also investigated the use of magnetic resonance imaging (MRI) in detecting skeletal abnormalities not seen on a skeletal survey; time to progression to myeloma was much shorter in patients with focal lesions on the MRI23 and these patients should be treated.
A number of models estimating the risk of progression to myeloma have been proposed. The presence of serum M-protein of greater than 3 g/dL, an FLC ratio outside the reference range of 0.125 to 8, and greater than 10 percent plasma cells in the marrow represents SMM with a high-risk of progression. Patients with these three risk factors had a cumulative risk of 76 percent of progression to myeloma within 5 years.15,24 The risk of progression decreased to 51 percent in patients with two of the risk factors and to 25 percent in patients with a single risk factor.24,25 Another clinical risk stratification model uses flow-cytometry of marrow aspirates. Using as risk factors (1) greater than 95 percent of all plasma cells being aberrant at diagnosis, (2) DNA aneuploidy, and (3) immune paresis, the presence of one, two, or three risk factors translated to 4, 46, and 72 percent risk of progression to myeloma with 5 years of observation, respectively.16 Other researchers have found that in addition to the intrinsic, molecular, and cytogenetic abnormalities of the plasma cells, an angiogenic switch and immunologic factors play a key role in the transformation of SMM to established myeloma.26
The development of myeloma is a complex multistep process involving karyotypic instability, Ig translocations, cell-cycle abnormalities (cyclin Ds), and multiple other mutations27 (Chap. 107). No molecular or chromosomal abnormalities can distinguish among monoclonal gammopathy, SMM, or myeloma at the time of diagnosis. Certain mutations occur in much higher frequencies in myeloma, such as p53 deletions, especially in refractory and extramedullary presentations,12,28 N-RAS and K-RAS mutations, chromosome 1p deletion and gain of 1q21, and translocations involving MYC (8q24).29,30,31,32,33 In whole-exome sequencing studies, intraclonal heterogeneity has been shown to be present at all stages of development—from monoclonal gammopathy to SMM to progressive myeloma.39 Genetic complexity increases as the disease progresses to myeloma.
Effect of Endogenous Factors There is an increased relative risk of monoclonal gammopathy and myeloma in overweight and obese patients based on their body mass index (BMI). With the exception of elite athletes, a BMI of 25.0 to 29.9 kg/m2 and 30 kg/m2 or greater define overweight and obese individuals, respectively.35,36,37,38,39,40,41 Fat tissue is a dynamic endocrine organ, secreting adipokines, hormones that play an important role in energy homeostasis and inflammation. Several adipokines, such as leptin and adiponectin, have been implicated in the development of cancer.40 Adiponectin levels are inversely correlated with obesity. Adiponectin serum concentrations were lower in monoclonal gammopathy patients who subsequently developed myeloma. In the KaLwRij strain of C57 black mice, which is permissive to the growth of 5T myeloma, compared to the parental strain of C57B16, which is not, adiponectin gene expression is significantly lower than in the parental strain. An increased myeloma burden was found in adiponectin-deficient mice, while pharmacologic enhancement of circulating adiponectin resulted in apoptosis of myeloma cells and also prevented bone disease. Fat tissue is a principal source of interleukin (IL)-6, one of the principal growth and antiapoptotic cytokines acting on myeloma cells. Obese individuals have been shown to have shorter telomeres than nonobese individuals. Because telomeres protect chromosomes from injury, including undesirable translocations, this effect may also contribute the relationship of body mass with PCN.
Effect of Exogenous Factors Aspirin has been shown not only to reduce cancer incidence, but to also dramatically decrease cancer mortality, especially in colorectal cancer, esophageal, gastric cancer, breast cancer, prostate cancer, and lung cancer. Aspirin inhibits several pathways that are important in myeloma, including nuclear factor κB (NF-κB), AKT activation, and the BCL-2 family of proteins. Aspirin is used frequently as thromboprophylaxis in myeloma patients receiving immunomodulatory therapy. In a prospective study designed to examine whether regular aspirin use influences the risk of myeloma, participants taking 5 or more tablets of 325 mg per week had a 39 percent lower myeloma incidence than nonusers. The association appeared stronger in men than in women.43 Aspirin inhibits proliferation and induces apoptosis of myeloma cell lines in vitro through regulation of BCL-2 and BAX and suppression of vascular endothelial growth factor (VEGF). In addition, in vivo studies in mice showed that aspirin administration resulted in retardation of tumor growth and in increased survival.44 A number of case-control and cohort studies have established that smoking has no association with the incidence of myeloma. Convincing evidence has not been found linking alcohol consumption to myeloma development.45
Occupation Many studies evaluated the potential role of exposure to certain occupations and/or toxin and the subsequent risk of myeloma development. Agricultural workers and farmers were studied in the United States and Europe. The majority of studies report an increase frequency of myeloma in agricultural workers, whereas other reports fail to find such a correlation.46 Exposure to toxins such as organic solvents (e.g., toluene, benzene), pesticides, paints, and others products with trace benzene content have been investigated for an association with the incidence of myeloma, but the findings are inconsistent.47
Radiation Studies on the survivors of the atomic bombing in Japan have failed to establish a cause-and-effect relationship between high-dose radiation exposure and an increased incidence of myeloma.48 Studies from the United Kingdom have reported no increased frequency of myeloma in workers exposed to ionizing radiation, nuclear plants, and/or plutonium.45 In a large study in China of x-ray technicians, there were no reported cases of myeloma or plasma cells disorders.49 Thus, radiation exposure is not linked to the risk of myeloma.
Chronic immune stimulation has not been shown to play a causative role in the etiology of myeloma. No link between infections, allergic conditions, or immunizations and the development of myeloma has been established. Patients with autoimmune disorders, in general, have not been found to have an excess risk of myeloma. Some studies report an increased risk of myeloma in HIV50,51 and hepatitis C patients,52 although more convincing data are needed to establish a cause-and-effect relationship.
Etiology In a small study of 65 patients and 213 controls, a preceding autoimmune condition did not predict the development of Waldenström macroglobulinemia.53 In contrast, many other studies have found such a link. In a large population-based study, that included 146,394 hepatitis C patients and 572,293 controls, a threefold increased risk of macroglobulinemia was observed along with a 20 to 30 percent increased risk of lymphoma.54 In a study of 361 U.S. veterans with Waldenström macroglobulinemia after a 27-year followup, a two- to threefold increased risk of developing the disease was found in patients with autoimmune-related conditions, hepatitis, HIV infection, and rickettsiosis.55 In two Swedish population-based studies, a personal history of autoimmune conditions and infections was associated with an increased risk of macroglobulinemia.56
Case-control studies and a large population-based study in Sweden have established the role of familial clustering of macroglobulinemia, thereby raising the concept of common susceptibility genes that could predispose to the disease.53,57,58,59,60,61 In study of macroglobulinemia, 19 percent of patients had at least one identified first-degree relative with macroglobulinemia or a B-cell disorder.60 In genome-wide linkage analyses of high-risk families with macroglobulinemia and IgM monoclonal gammopathy, the strongest linkage was found to be chromosomes 1q, 3q, 4q, and 6q.58,60,62,63,64 In a gene-sequencing study performed on marrow cells of macroglobulinemia patients, a recurrent somatic mutation of the gene MYD88L265P on chromosome 3 that encodes signal transduction and innate immunity was found to be in approximately 91 percent of the patients tested.60
In a novel study using expression cloning, several common self-antigens designated Paratarg-7 were discovered.10 When carriers of the phosphorylated forms of Paratarg-7 were studied, they were found to have a 6.5-fold higher risk of developing IgM monoclonal gammopathy or macroglobulinemia. The antigen causes continuous autostimulation of cognate T-helper cells, which, in turn, specifically activate B cells with high affinity to Paratarg-7.
GENETIC ABNORMALITIES IN PLASMA CELL NEOPLASM
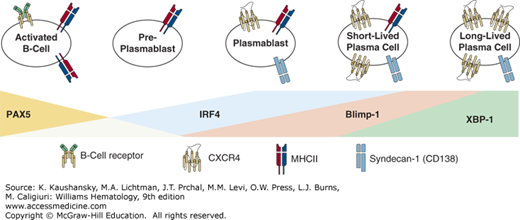
Plasma cell differentiation begins in lymph nodes and in the spleen where cells undergo changes in gene expression and cell-surface molecules followed by migration to the marrow or mucosal lamina proper.65 The development of plasma cells alters the cellular receptor landscape of the cell with the deletion of important B-cell receptors and the addition of receptors necessary for plasma cell function and antibody production. Changes to cellular receptors include downregulation of major histocompatibility complex (MHC) class II, CD19, CD21, and CD22.66 Perhaps, the most important alterations during myeloma development are decreases of the B-cell receptor (BCR), CXCR5, and CCR7. In contrast, plasma cells upregulate CXCR4, CD138, and CD38 (Fig. 105–1).66,67,68 Plasma cells also undergo changes to transcription factors highlighted by a decrease in PAX5, CIITA, and EBF.66,67,68,69,70 Furthermore, plasma cells express genes that are present in B cells at low levels or not at all and are highlighted by increased expression of Blimp-1, IRF4, and XBP1, the only transcription factor exclusively required for plasma cell development.71 For more information on plasma cell development (Chap. 74).
Figure 105–1.
Overview of plasma cell differentiation. The differentiation of a plasma cell from a B-cell lineage occurs over a multistep process using plasmablast and short-lived plasma cell intermediates. Throughout the process, there is a profound change in gene expression and cell-surface markers, which allows for the identification of each stage based on the cell-surface marker expression and gene expression of transcription factors important to plasma cell differentiation. Depicted here are necessary changes to transcription factors and cell-surface markers that occur during each phase of plasma cell differentiation.
Early genetic events in myeloma genesis include the accumulation of sequential genetic changes; however, the full mechanism remains elusive.72 Protein kinases provide selective growth advantages to cells and, therefore, act as driver mutations, or inducers of early neoplastic events.73 The dysregulation of the cyclin D genes exposes cells to additional proliferative stimuli and commonly occurs as a result of a translocation of the cyclin D gene to the Ig loci.74 Activation-induced deaminase contributes to genetic instability through its involvement in induction of somatic mutations in the immunoglobulin genes and immunoglobulin translocations.75 Although important, gene alterations are not the only driving force in the development of PCN. Although ethnicity and genealogy affect the prevalence of monoclonal gammopathy,76 they do not impact the rate of progression from monoclonal gammopathy to myeloma. Furthermore, there is a higher incidence in monoclonal gammopathy in relatives of myeloma patients than seen in the general public.77,78 These results demonstrate that not only are there somatic mutations important to the development of PCNs, but genetic background also contributes to the likelihood of the development of plasma cell diseases.
Much focus has been placed on risk factors and early genetic events that can be used as diagnostic and prognostic markers. However, we are learning more about the genetic alterations that occur throughout disease progression. One of the more prominent discoveries focuses on the activation of NF-κB. Approximately 50 percent of myeloma patients exhibit canonical NF-κB activation.79,80 NF-κB functions, in part, by regulating growth and survival within the myeloma cells; overexpression of positive regulators, such as NIK, NFKB1, NFKB2, and CD40, and inactivation of negative regulators, such as CLYD, TRAF2, TRAF3, and cIAP1/cIAP2, contribute to the constitutive activation of NF-κB within these cells.80
RAS mutations are also important contributors to the development of myeloma from monoclonal gammopathy. Oncogenic activation of RAS occurs through mutation of one of three different codons with mutations resulting in constitutively activated RAS. Less than 5 percent of cases of monoclonal gammopathy display mutations of RAS, whereas in newly diagnosed myeloma patients mutated RAS is found in nearly 40 percent of patient samples, suggesting that RAS may be associated with the conversion of monoclonal gammopathy to myeloma.81,82,83
p53, which regulates the cell cycle and acts as a tumor suppressor represents the most commonly inactivated tumor-suppressor gene in cancer.84,85 In newly diagnosed PCNs, p53 mutations occur in 5 percent of patients. The frequency of p53 mutations increases with disease progression; while infrequent in newly diagnosed myelomas, 30 percent of plasma cell leukemia patients present with p53 mutations.86,87 Furthermore, p53 mutations are negatively correlated with survival.88 Similar in function to p53, the retinoblastoma (RB) gene regulates the cell cycle. RB functions by inhibiting the effects of the cyclin D proteins with the help of the p18INK4 proteins. However, overexpression of cyclin D or decreased expression of RB can lead to cell-cycle progression and neoplastic growth. Decreased expression of the two RB pathway regulators p18INK4a and p18INK4c can inhibit the regulatory effects of RB and result in cell-cycle release. Decreased expression of the two proteins is considered to be a late disease progression event.76
Myeloma is genetically heterogeneous and more closely resembles solid tumors than other hematologic malignancies. Because of its high degree of tumor heterogeneity, myeloma gene-expression microarrays have proven invaluable to our understanding and treatment of myeloma. Four stratification models exist designed to classify myeloma based on the risk profile as determined by gene-expression profiles.89 Of the four, one model has proven most reliable, as it retains its prognostic relevance with newer therapeutic regimens. This model compares the expression of 70 different genes to devise a scoring system that ranks a sample as high risk (13 percent of patients) or low risk.90 Abnormalities of expression that map to chromosome 1 are disproportionately represented within the model, with an impressive number of upregulated genes mapping to 1q and a high percentage of downregulated genes mapping to 1p. The 70-gene model offers a reliable method for the detection of high-risk myeloma. However, it does not take into account all important genes related to myeloma. One important gene not found in this model is MYC, which is a transcription factor that influences the expression of many genes through the binding of consensus sequences within the noncoding region of genes. It is thought to regulate the expression of 15 percent of all genes.91 MYC expression within myeloma and monoclonal gammopathy varies depending on the state of the disease. In high-risk monoclonal gammopathy and early stages of myeloma, the increase in MYC expression is primarily a result of a decrease in transcriptional regulation, whereas in late disease states, the 8q region encoding MYC translocates to the immunoglobulin loci, resulting in dysregulation of expression because of the influence of neighboring regulatory elements.32
Refractory myeloma has several causes, with dysregulation of gene expression contributing significantly to the overall development of drug resistance. The serine/threonine kinase NEK2 induces drug resistance in myeloma through the activation of efflux drug pumps.92 Overexpression of NEK2 results in the activation of the AKT pathway and subsequently of NF-κB, which results in the upregulation of ABC drug efflux transporters. Overexpression of the antiapoptotic molecule MCL-1 induces drug resistance through the inhibition of apoptosis.93 More specifically, MCL-1 overexpression results in the inhibition of the proapoptotic BCL-2 family members, which results in blocking apoptosis.
Next-generation sequencing or deep sequencing relies on the ability to sequence large amounts of small DNA fragments quickly and assemble the output into a complete coherent data set. This technique is providing new insights into our understanding of existing neoplastic mechanisms and identifying novel mechanisms and genes that had evaded detection thus far. One of the first reports to use next-generation sequencing was from a study that compared 38 whole-tumor genomes from patients with myeloma to matched normal DNA.94 Novel genes involved in histone methylation, protein translation and blood coagulation were identified. Mutations to the toll-like receptor (TLR) 4, the adaptor molecule TRAF3, and to the kinases CYLD, RIPK4, and BTRC also were discovered. A greater-than-anticipated change in NF-κB was observed, including identifying 11 mutated genes involved in the NF-κB pathway. Furthermore, the prognostic value of next-generation sequencing was tested by using the sequencing technology to detect minimal residual disease (MRD) in myeloma patient samples.95
Fluorescence in situ hybridization (FISH) has become the gold standard for the detection and classification of myeloma. However, FISH cannot provide information about chromosomal abnormalities without the use of large scale panels of probes. Comparative genomic hybridization (CGH) arrays overcome some of the short falls of FISH technology by providing a genome-wide view of chromosomal changes, but do so at a reduced resolution of 10 to 20 megabases. To combat the low resolution of CGH arrays, small nucleotide polymorphism (SNP)-based technology has been employed. SNP arrays can detect copy number changes and have improved the resolution of CGH arrays to a submegabase level. An early report validated the use of SNP arrays by comparing its results to those obtained by FISH analysis using identical samples. Furthermore, uniparental disomy (UPD) was identified as prevalent in myeloma samples and may occur through several mechanisms, including mitotic nondisjunction and mitotic recombination.96 A technical advance was the introduction of the Affymetrix Cyto Scan HD arrays. The Cyto Scan arrays incorporate the most up-to-date SNP library to generate a 2.6-million probe library across the human genome, allowing for resolution up to 50 kb. The technology has not been published in a myeloma study yet, but the resolution matches that of FISH and can provide a wealth of knowledge related to changes in copy number, mosaic chromosomes and loss of heterozygosity. Using these technologies numerous cytogenetic changes were identified that may be important for the development and progression of monoclonal gammopathy to myeloma.









