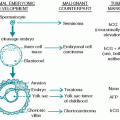
I. EPIDEMIOLOGY AND ETIOLOGY
A. Classification of diseases associated with monoclonal paraproteinemia
1. Plasma cell neoplasms
a. Multiple myeloma (MM)
b. Amyloidosis
c. Heavy chain disease
d. Papular mucinosis
2. Other neoplastic diseases
a. Waldenström macroglobulinemia (WM)
b. Malignant B-cell non-Hodgkin lymphoma, chronic lymphocytic leukemia (CLL)
c. Neoplasms of cell types not known to synthesize immunoglobulins (solid tumors, monocytic leukemia, myelodysplastic syndromes)
3. Nonneoplastic disorders
a. Monoclonal gammopathy of undetermined significance (MGUS)
b. Autoimmune diseases (e.g., systemic lupus erythematosus)
c. Hepatobiliary disease
d. Chronic inflammatory diseases
e. Immunodeficiency syndromes
f. Miscellaneous diseases (e.g., Gaucher disease)
g. Pseudoparaproteinemia (see Section IX.C)
B. Incidence. MGUS, MM, and WM are the most common disorders associated with M-proteins. The average age at the time of diagnosis is 65 years, and the incidence increases with age.
1. MGUS (formerly benign monoclonal gammopathy). The approximate incidence of MGUS is 0.2% for patients 25 to 49 years of age, 2% for those 50 to 79 years of age, and 10% for those 80 to 90 years of age.
2. MM develops in 3/100,000 population and constitutes 1% of new cancer cases in the United States. The average age is 65 years and many patients are >70 years of age. Men are affected slightly more often than women. MM is the most common lymphohematopoietic malignancy in blacks.
3. WM has an incidence that is about 5% to 10% of that of MM. Two-thirds of cases occur in men.
4. Lymphomas. Excluding MGUS, MM, and WM, about half of the patients with monoclonal gammopathies have lymphocytic lymphoma or CLL. The M-protein is nearly always either IgM or IgG and usually causes no symptoms. Patients with other types of lymphoma do not have an increased incidence of monoclonal proteins.
C. Etiology. No specific etiologic agent for the plasma cell dyscrasias has been found. Predisposing factors in humans appear to be the following:
1. Radiation exposure increases the risk of MM. Survivors of the atomic bomb in Japan have been shown to have a higher risk of developing monoclonal gammopathies.
2. Chronic antigen stimulation. Many M-proteins have been shown to be antibodies directed against specific antigens, such as microbial antigens, red blood cell antigens, neural antigens, lipoproteins, rheumatoid factors, and coagulation factors. Chronic antigenic stimulation (e.g., chronic osteomyelitis or cholecystitis) may predispose to the development of MM or MGUS. Patients with autoimmune disease may be at high risk for MM. A recent case-controlled study suggests that female patients who have had silicone gel breast implants also may be at high risk for MM.
3. Environmental exposure. Exposure to pesticides and benzene in the workplace and the use of hair dye are associated with an increased incidence of MM. Farmworkers have been shown to be at high risk for MM in several epidemiologic studies.
4. Human herpesvirus 8 (HHV-8) has been found in the nonmalignant bone marrow dendritic cells of patients with myeloma. It remains to be determined if HHV-8 contributes to the growth of the malignant plasma cells in these patients.
5. Family history of a monoclonal gammopathy is a risk factor for the development of a plasma cell dyscrasia.
D. Cytogenetics
1. MM. Multiple, complex karyotypic changes are observed in the malignant plasma cells of most patients. Fluorescent
in situ hybridization (FISH) analysis has shown that most patients with MM have malignant cells with translocations involving chromosome 14 at the site of the immunoglobulin heavy chain gene locus and a limited number of nonimmunoglobulin
partner chromosomes. Unlike the site of translocation in other B-cell malignancies that involves the joining region JH, the location of the breakpoint in myeloma usually occurs in the switch regions that are involved in heavy chain class switching from Cµ to another heavy chain class.
Hyperdiploidy is observed in approximately 40% of cases of MM and associated with an improved survival. In contrast, patients with hypodiploidy have a worse outcome as this is commonly associated with chromosomal translocations associated with poor survival.
a. The most common sites of the nonimmunoglobulin breakpoints include chromosomes 11 at the site of cyclin D, chromosome 16 at the site of the c-MAF proto-oncogene, and chromosome 4 at the site of the fibroblastic growth factor receptor 3. Loss of material on the long arm of chromosome 13 occurs in nearly 20% of patients. Loss of 17p is observed in approximately 10% of patients. In addition, addition of material at 1q21 commonly occurs in MM.
Prior to bortezomib- and lenalidomide-based therapies (see below), specific translocations such as 4;14 were associated with poor outcomes, whereas patients with 11;14 translocations had improved outcomes. However, following the introduction of these agents in combination therapies, only chromosome 17p loss is associated with poor survival.
b. Mutations of ras genes occur in about 20% of myelomas and are associated with a poor prognosis. Similarly, mutations of p53 are found in 15% to 20% of cases and are associated with more advanced and clinically aggressive disease. Abnormalities in the c-MYC proto-oncogene may occur much more commonly than was previously suggested.
These studies have led to a new classification of myeloma based on these results. Recent studies suggest that clinical features such as the presence of bone disease are associated with specific gene-expression profiles. In addition, single-nucleotide polymorphism and comparative genomic hybridization studies have identified patients with different outcomes in MM.
c. Gene-expression profiling has identified specific subgroups of patients with MM. These studies have suggested a markedly different outcome depending on the expression of specific genes. These profiles also may predict responsiveness to specific therapies.
d. Telomerase activity and telomere length, indicators of the aging of specific cells, are directly related to the type of myeloma as well as to the outcome among patients. Patients with higher telomerase activity and shorter telomere length tend to have a poor prognosis.
2. MGUS. Studies have shown that patients with MGUS have similar karyotypic abnormalities to patients with MM.
3. WM. Complex karyotypes are also commonly observed in WM. Occasional patients have translocations involving the immunoglobulin heavy chain locus on chromosome 14 and either c-MYC on chromosome 8 or BCL-2 on chromosome 18.
II. PATHOLOGY AND NATURAL HISTORY
A. Bone marrow pathology is usually distinctive in MM and WM. Plasma cells that constitute ≥10% of the nucleated marrow cells (excluding erythroblasts) are characteristic but not diagnostic of MM.
1. MGUS. By definition, patients have <10% light chain-restricted plasma cells.
2. MM. Plasma cells usually constitute 10% to 100% of the marrow cells; they have abundant basophilic cytoplasm and eccentric nuclei with paranuclear clear zones. Immaturity of the plasma cells is evident with the presence of prominent nucleoli (“myeloma cells”). Bone marrow biopsy showing monotonous infiltration with plasma cells is the only diagnostic criterion for MM accepted by many authorities. The presence of large, homogeneous infiltrates or nodules of plasma cells is highly suggestive of MM. Early in its course, however, marrow involvement is patchy, and normal marrow particles may be obtained.
3. WM may closely resemble CLL. Bone marrow in WM contains 10% to 90% plasmacytoid lymphocytes or small mature lymphocytes; mast cells are often prominent.
4. Reactive plasmacytosis. Peripheral blood plasmacytosis occurs in many viral illnesses (including human immunodeficiency virus [HIV] infection), serum sickness, and plasma cell leukemia (which is rare). Bone marrow plasmacytosis, when not caused by myeloma, is characterized by a diffuse distribution (not infiltrative) and alignment of mature plasma cells along blood vessels or near marrow reticulum cells. Since reactive plasmacytosis is not a malignant clonal disorder, the plasma cells do not show kappa or lambda light chain restriction. Reactive bone marrow plasmacytosis is commonly seen in many disorders, including the following:
a. Viral infections
b. Serum sickness
c. Collagen vascular disease
d. Granulomatous disease
e. Liver cirrhosis
f. Neoplastic disease
g. Marrow hypoplasia
B. Natural history of MGUS. MGUS occurs in nearly 5% of individuals over the age of 70. Although these individuals are symptom-free at diagnosis, nearly 25% of cases progress to a malignant disorder (usually MM) by 25 years of follow-up.
Importantly, the risk for malignancy remains constant over time (approximately 1%/year). The risk of developing MM from MGUS is directly related to the size of the monoclonal peak. The presence of a high ratio of abnormal to normal plasma cells as characterized by immunofluorescence has been shown to predict a higher risk of developing MM. Some studies suggest that patients with MGUS are at higher risk of developing accelerated bone loss and fractures, especially of the vertebral bodies. In addition, these patients appear to be at higher risk of developing thromboembolic events.
1. Karyotypic abnormalities in these patients are similar to those seen with MM.
2. The presence of depressed normal immunoglobulin levels occurs in many patients with MGUS, but is not associated with a higher risk for infection and does not predict a higher risk for malignancy.
3. Peripheral neuropathy is not uncommon and may be associated with a monoclonal antibody with reactivity to a myelin-associated glycoprotein (see Section IX.B).
C. Natural history of WM. WM originates from clones of lymphocytes or plasma cells that synthesize µ chains. The natural history of WM resembles lymphocytic lymphoma much more than MM. Many patients do not require therapy for more than a decade of follow-up. Separating WM from
MGUS, CLL, or lymphocytic lymphoma with IgM spikes may be more arbitrary than real.
Lymphadenopathy, splenomegaly, and hyperviscosity are hallmarks of WM; skeletal lesions and impaired renal function are unusual. Concomitant macroglobulinemia and osteolytic lesions usually signify malignant lymphoma or solid tumor rather than primary WM. Glomerular lesions are frequent in WM, but renal failure is uncommon. Low levels of light chains in the urine occur in about 25% of patients.
D. Natural history of MM. Three to twenty years of clonal growth may pass before MM becomes clinically evident. The disease may be localized (5% of cases), indolent (10%), or disseminated and progressive (85%). Nearly all cases of MM originate as MGUS. Manifestations of disease progression arise from bone marrow and skeletal involvement, plasma protein abnormalities, and the development of renal disease.
1. Hematopoiesis is often impaired. At the time of diagnosis, 60% of patients have anemia; 15%, leukopenia; and 15%, thrombocytopenia. Nucleated red blood cells and immature granulocytes may be present in the peripheral blood (leukoerythroblastic reaction).
2. Plasmacytomas (plasma cell tumors) may develop anywhere in the skeleton or, rarely, in extraskeletal sites, such as the nasopharynx or paranasal sinuses. Localized plasmacytomas produce a monoclonal spike in the serum or urine protein electrophoresis in only half of the cases. The median survival is >8 years. Most plasmacytomas that appear to be solitary become generalized in about 3 years, particularly those involving the skeleton. Extraskeletal plasmacytomas have a better prognosis than those of skeletal origin and less frequently progress to multiple myeloma.
3. Skeletal disease in MM
a. Osteolytic lesions. Multiple osteolytic lesions are present in about 70% of patients at diagnosis, single osteolytic lesions or diffuse osteoporosis in 15%, and normal skeletal radiographs in 15%. Lesions are most commonly seen in the skull, vertebrae, ribs, pelvis, and proximal long bones. The use of MRI indicates that skeletal abnormalities exist in nearly all patients with myeloma.
Previously, it was thought that the demineralization and lytic lesions occur as a result of osteoclastic-activating factors and osteoblasticinhibitory factors produced by neoplastic plasma cells and activated by inflammatory cytokines. The loss of bone in these patients, however, now appears to be a complex interplay involving the tumor cells, stromal cells in the bone marrow, and both the osteoblasts and osteoclasts. The factors responsible involve other important molecules, including macrophage colony-stimulating factor, vascular endothelial growth factor, specific matrix metalloproteinases, macrophage inflammatory protein-1 α (MIP1-α), dickkopf1 (DKK-1), secreted frizzled protein-3, and the receptor for activation of nuclear factor-κB (NF-κB). The latter receptor is designated as “RANK” and is coupled with RANK ligand (RANKL) to comprise the RANKL-RANK signaling pathway.
(1) RANK-RANKL proteins play a key role in the development of myeloma bone disease. Increased levels of RANKL have been found in myeloma bone marrow and are associated with enhanced bone loss.
(2) Osteoprotegerin (OPG), the natural soluble decoy inhibitor of RANKL-RANK signaling, is decreased in MM bone marrow and
blood. Blockade of RANKL prevents skeletal lesions in animal models of MM. The ratio of circulating RANKL/OPG predicts bone disease in MM patients.
(3) The chemokine macrophage inflammatory protein (MIP1-α) also appears to play a key role in myeloma bone disease. MIP1-α is elevated in myeloma bone marrow; it is associated with increased bone loss and may stimulate myeloma cell growth.
(4) DKK1, an inhibitor of osteoblast development and function, also has an important role in myeloma bone disease. Levels of DKK1 are elevated in the blood and bone marrow from patients with myeloma compared with normal subjects. The inhibition of osteoblast function ultimately leads to a loss of bone formation and enhanced bone loss.
b. Osteoblastic lesions occur in <2% of patients, often in association with neuropathy and the POEMS syndrome. Because of their rarity, the diagnosis of MM should be doubted in the presence of osteoblastic lesions.
c. POEMS syndrome is a multisystem disorder usually associated with osteosclerotic myeloma. It is characterized by the combination of polyneuropathy (chronic inflammatory demyelinating neuropathy), organomegaly, endocrinopathy, M-protein (mainly IgG-γ or IgA-γ), and skin changes (hyperpigmentation, thickening, hypertrichosis). Various other signs, such as cachexia, fever, edema, clubbing, and telangiectasia, can also occur. Autoantibodies to peripheral nerve components are absent. The syndrome appears to be the result of marked activation of the proinflammatory cytokines. Patients with POEMS syndrome, particularly those associated with Castleman disease, have been found to contain HHV-8.
d. Hypercalcemia. About 10% of patients with MM present with hypercalcemia, and 10% develop it during the course of their disease. This complication results from enhanced bone resorption, resulting in the release of calcium into the circulation. Hypercalcemia is a major cause of renal failure among patients with MM, and normalization of the serum calcium often reverses the renal dysfunction. Avoid bed rest and immobilization because these factors can contribute to both the development and worsening of hypercalcemia. Serum alkaline phosphatase levels are usually normal but may be increased with recalcification of fractures. It is important to remind patients who are on calcium and vitamin D supplements to discontinue these supplements until the calcium level is under control.
4. Protein abnormalities
a. Frequency. The incidence of monoclonal immunoglobulins in MM and in comparison to MGUS is shown in
Table 22.2.
b. Increased excretion of κ or λ light chains in the urine depends on the rate of unbalanced synthesis of excess light chains, plasma volume, degradation rate, renal catabolism, and urine volume. Monoclonal light chains in the urine are present in two-thirds of all patients with MM and present without an M-protein in the serum in 25%.
c. Serum free light chains are identified in MM patients and, importantly, in many patients with otherwise “nonsecretory” disease.
d. Normal immunoglobulins are usually decreased in the serum of patients with MM and are occasionally decreased in patients with MGUS. The mechanism of inhibition of their synthesis is unknown. Older series showed a high rate of infection with encapsulated organisms that was thought to be related to patients’ marked decrease in normal serum immunoglobulins. The risk for infection, however, largely occurs during chemotherapy-induced neutropenia or during the terminal stages of the disease.
e. Other plasma alterations (see Section IX.A). Hyperviscosity is unusual in MM (<5% of patients).
5. Renal dysfunction, both acute and chronic, occurs at diagnosis in 15% to 20% of cases and develops during their course in most patients with MM. Many patients have renal dysfunction from causes other than MM owing to comorbid diseases such as diabetes mellitus or hypertension, urinary tract infections, nephrotoxic medications, or dehydration. Patients with MM secreting urinary light chains commonly present with renal failure. The most important causes of renal dysfunction in these MM patients are hypercalcemia and myeloma kidney.
a. Myeloma kidney is generally attributed to the deposition of κ and λ chains in the distal and collecting tubules, which is where the light chains are catabolized. The tubules dilate, apparently obstructed by casts surrounded by multinucleated giant cells, and undergo cellular atrophy. Glomerular basement membrane disease also occurs in most patients with myeloma kidney. In most instances, proteinuria contains monoclonal light chains only. These abnormalities occur slightly more commonly in MM associated with λ chain production.
Malignant myeloma is the most common cause of the adult Fanconi syndrome (aminoaciduria, glycosuria, phosphaturia, and electrolyte loss in the urine). Fanconi syndrome may precede the recognition of MM by many years.
b. Amyloidosis also develops commonly in MM. It affects the glomeruli and results in nonselective proteinuria.
c. Inconstant findings that may aggravate renal function include pyelonephritis, metabolic abnormalities in addition to hypercalcemia (nephrocalcinosis and hyperuricemia), glomerulosclerosis, and focal myeloma cell infiltration. Renal tubular acidosis occasionally occurs. Nephrotic syndrome is rare in MM unless amyloidosis supervenes. Recent studies suggest, however, that chronic administration of IV pamidronate may also be associated with nephrotic syndrome (see below).
d. Intravenous contrast-dye studies should be done with caution (if at all) because patients with MM are more susceptible to renal dysfunction after such studies, particularly if they are dehydrated.
6. Neurologic dysfunction often develops in MM and is the result of several pathogenetic mechanisms.
a. Central nervous system (CNS). Spinal cord and nerve root compression develops in about 15% of patients and is usually caused by epidural
plasmacytoma. Amyloidosis is a rare cause of epidural masses. Collapse of vertebral bodies can also cause spinal cord compression but, more likely, produces radicular symptoms secondary to nerve root compression. Cranial nerve palsies can develop from tumor occlusion of calvarial foramina. Intracerebral and meningeal plasmacytomas are rare.
With the longer survival of myeloma patients, meningeal myeloma seems to be occurring more frequently than had been previously observed. Furthermore, many of the newer anti-MM agents are recognized to cause fatigue and cognitive dysfunction.
b. Peripheral neuropathy. The carpal tunnel syndrome, which is usually the result of amyloid infiltration of the flexor retinaculum of the wrist (causing entrapment of the median nerve), is a common peripheral neuropathy in MM. Infiltration of nerve fibers and vasa nervorum with amyloid can also produce peripheral neuropathy. Additionally, peripheral neuropathy may be associated with monoclonal immunoglobulins to myelin-associated glycoproteins (see Section IX.B). Rarely, patients with MM and POEMS syndrome develop a characteristic peripheral neuropathy. The most common cause of peripheral neuropathy in patients with MM is from treatment with drugs such as thalidomide, bortezomib, or arsenic trioxide.
c. Neurologic paraneoplastic syndromes (see
Chapter 32, Section V)