Plasma Cell Disorders
Tomer M. Mark, MD
Geraldine P. Schechter, MD, MACP
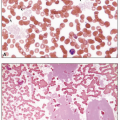
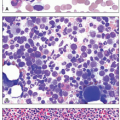
Plasma cells are terminally differentiated B cells responsible for immunoglobulin production. Plasma cell disorders represent a group of malignancies in which clonal plasma cells give rise to a spectrum of pathology, depending on the degree of plasmacytosis in the bone marrow, the amount and type of immunoglobulin secretion, and the tropism of the immunoglobulin deposition in the body.1 The appearance of the plasma cells in the bone marrow may vary widely in these neoplasms, ranging from similar to normal mature plasma cells to very immature plasmablastic cells with high nuclear:cytoplasmic ratios. Abnormal plasma cells may produce immunoglobulin to such a degree that vacuoles full of immunoglobulin can either aggregate in the cytoplasm, in which case they are called Russell bodies, or penetrate the nucleus, then termed Dutcher bodies. A subset of immunoglobulins can also produce a highly eosinophilic appearance to the plasma cell cytoplasm, especially in the case of monoclonal IgA isotype antibodies, thereby giving rise to the term “flame cell.”2
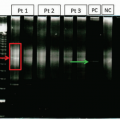
Plasma cell disorders are characterized by secretion of a monoclonal immunoglobulin (either whole or light chain only; very rarely heavy chain only) which can be quantified by serum protein electrophoresis (SPEP). The monoclonal immunoglobulin is referred to as an M-spike (because of the characteristic peaked appearance of the γ region on the densitometry scan of an electrophoresis gel), a monoclonal paraprotein, or an M-protein. The M-spike isotype is determined via immunofixation, in which parallel SPEPs in a gel are each impregnated with a specific antibody to the heavy chain of immunoglobulin IgG, IgA, or IgM and the light chains, κ or λ. The presence of an M-protein will produce a distinct pattern on the immunofixation gel correlating with the isotype.3
Clonal plasma cells often produce excess free κ- or λ-light chains which are not bound to heavy chains, in addition to the intact M-protein.4 These circulate in the blood and are excreted via the kidneys. Free light chains are normally reabsorbed in the proximal tubule of the nephron; however, when in great excess, they can be found in the urine and are then called Bence Jones proteins. Similar to serum testing, a Bence Jones protein can be identified by urine protein electrophoresis and immunofixation. Free light chains in the serum can also be measured by an immunologic assay using antibodies that will detect only free, and not bound, light chains.5
The presence of an M-spike that has been confirmed to have a single heavy and/or light chain by immunofixation is referred to as a monoclonal gammopathy. Although monoclonal gammopathy is most often linked to plasma cell neoplasms, there are many hematologic and immunologic disorders in which a monoclonal gammopathy can also be present. These conditions range in clinical impact from benign diseases (such as monoclonal gammopathy of undetermined significance [MGUS], rheumatologic diseases, skin disorders, chronic viral infections) to malignancies of B cells, including multiple myeloma (MM), Waldenström macroglobulinemia (WM), and B-cell lymphomas (Table 10.1).6 In the case of connective tissue disorders, the circulating autoantibody associated with the disease, such as rheumatoid factor, can manifest as an M-spike on SPEP testing thus producing an associated monoclonal gammopathy.7
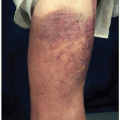
Monoclonal gammopathies may present as type I cryoglobulinemia. Type I cryoglobulinemia involves a monoclonal immunoglobulin, usually of the IgG or IgM subtype, that precipitates from serum and plasma at temperatures below 37°C. The most common sources of these monoclonal immunoglobulin are WM, monoclonal plasma cell disorders, or other lymphoproliferative disease.8,9 Type I cryoglobulinemia may be asymptomatic or present with a constellation of signs and symptoms such as acrocyanosis, gangrene of distal extremities, hyperviscosity syndrome, skin purpura or ulceration, peripheral neuropathy, arthralgias, and membranoproliferative glomerulonephritis. Type II cryoglobulinemia results from a combination of a monoclonal protein with a polyclonal IgG and is associated with immune complex-mediated vasculitis often in conjunction with hepatitis C infection.10
There are two rare severe skin diseases that are highly associated with monoclonal gammopathy, although the pathophysiology is obscure and not usually associated with an underlying plasma cell disorder. Schnitzler’s disease, a neutrophilic urticarial dermatosis, is associated with monoclonal IgM-κ in over 75% of cases.11 This debilitating condition is characterized by the development of chronic nonpruritic urticarial lesions with signs of systemic inflammation, including periodic fevers and leukocytosis. Schnitzler’s disease has recently been discovered to respond to anakinra, an interleukin-1 receptor antagonist.12 Scleromyxedema is a form of papular mucinosis that is associated with IgG-λ monoclonal gammopathy in 70% to 90% of the cases.13 In this condition, papules filled with mucin form and coalesce on the skin, leading to induration and restriction of movement in a scleroderma-like process. Scleromyxedema responds to high-dose intravenous immunoglobulin with occasional responses to immunomodulating novel agents used to treat MM.
MONOCLONAL GAMMOPATHY OF UNDETERMINED SIGNIFICANCE
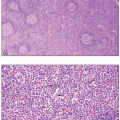
MGUS is the most common plasma cell disorder. MGUS arises from a limited clonal proliferation of plasma cells in the bone marrow which secrete M-protein but otherwise has no clinical complications. The World Health Organization (WHO) definition of MGUS is a serum M-spike of less than 3 g/dL associated with less than 10% monoclonal plasma cells in the marrow, the absence of end organ damage, and no evidence of a lymphoma, as many lymphomas can be associated with a monoclonal gammopathy incidentally. MGUS is common in the elderly, and its incidence increases with aging; approximately 3% of persons over the age of 50 and 5% of persons over the age of 75 in the United States have MGUS.14 There is an increased risk of persons with MGUS to progress to symptomatic MM of approximately 1% per year. The risk of progression is higher when there is a greater amount of M-protein present, a non-IgG isotype of M-protein, and an abnormal free κ:λ light chain ratio.15 Studies of sera collected prior to a new diagnosis of MM suggest that all cases of MM have a preceding MGUS condition.16,17
Monoclonal gammopathy of renal significance (MGRS) describes conditions where the M-protein, or portion thereof, harms the kidney.18 In these patients, low-level M-proteins have the capacity to damage glomeruli by a variety of mechanisms that lead to glomerulonephropathy and renal failure. In monoclonal immunoglobulin deposition disease, free light or heavy chains or both form extracellular non-organized deposits predominantly in the glomeruli and, in some patients in renal tubules as well, causing nephron damage manifested by Bence Jones proteinuria, heavy albuminuria, and renal insufficiency.19 The causative protein can often be readily determined by examination of the urine by immunoelectrophoresis and immunofixation, although renal biopsy with immunofluorescence is the gold standard for diagnosis. A bone marrow biopsy should also reveal a clonal population of plasma cells sharing the same isotype of monoclonal protein involved in the MGRS. Free light chains that are misfolded may also form organized aggregates in the renal parenchyma in the form of amyloid deposition, leading to nephrotic range albuminuria, in contrast to the Bence Jones proteinuria seen in light chain deposition disease. Renal failure is the most common manifestation of primary amyloidosis (discussed below); however, other organ damage caused by amyloid deposition is commonly seen. Rarely, monoclonal protein deposition has also been detected in renal biopsies from patients with proliferative membranous glomerulonephritis. Free light chains may also deposit on nerves and lead to a peripheral neuropathy in plasma cells disorders, including MGUS, MM, and POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) syndrome.20 A specific IgM antibody called myelin-associated glycoprotein (MAG) can cause demyelinating peripheral nerve damage.21
In free light chain deposition disease, extrarenal symptoms are rare; however, free heavy chain deposition disease often has other systemic morbidities depending on the heavy chain isotype. In the heavy chain diseases, mutated and truncated free heavy chains are unable to form disulfide bonds with corresponding light chains to create an intact immunoglobulin. Antisera will react with the corresponding heavy chain involved, but not with light chains. In α-chain disease, (also called Seligmann’s disease), the free α-chains have a predilection for deposition in the aerodigestive tract, where IgA is normally present in high amounts. Patients with α-chain disease are usually of Middle Eastern or Northern African descent and can present with symptoms of malabsorption or dyspnea, depending on whether the deposition involves primarily the gastrointestinal or respiratory tract. γ-Chain disease (also called Franklin disease) is less common than α-chain disease. γ-Chain disease occurs most frequently in conjunction with a history of autoimmune disease, such as rheumatoid arthritis, and more commonly affects women. γ-Chain disease can be diagnosed concurrently with a lymphoma in the majority of new cases and often involves Waldeyer ring. Fevers, lymphadenopathy, palatal edema, and impaired immunity are the most common symptoms.
MULTIPLE MYELOMA
MM is the second most common hematologic malignancy, representing approximately 10% of all blood cancers.22 MM is a chronic and incurable malignancy characterized by responses to treatment followed by relapses requiring retreatment.23 MM has classically been defined by the presence of all the following criteria: (1) at least 10% bone marrow clonal plasmacytosis or more than one plasmacytoma elsewhere in
the body; (2) detectable M-protein in the blood or urine; and (3) end organ damage resulting from the plasmacytosis.24 The end organ damage is often summarized by the acronym CRAB which stands for hypercalcemia, renal dysfunction, anemia, and bone lytic lesions. A recent update has been made to the definition of MM by the International Myeloma Working Group (IMWG) to define MM as the presence of clonal plasmacytosis in the marrow or a plasmacytoma plus one or more of the following biomarkers of malignancy: at least 60% bone marrow plasmacytosis, free light chain (κ:λ or vice versa) ratio of at least 100, or more than one focal lesion on a MRI study.25 The presence or lack of M-protein further defines the MM into secretory and nonsecretory types. Thus, a person may have a biomarker of malignancy without symptoms and still have the diagnosis of MM. The diagnosis of smoldering myeloma (SMM) is made when a person has a clonal marrow plasmacytosis and/or M-protein present yet does not otherwise fulfill other criteria for either MGUS or MM.26 Patients with SMM have an elevated risk of transformation to active MM at approximately 10% per year for the first 5 years from diagnosis, then 3% per year for the next 5 years, followed thereafter at a rate of 1% per year. Most patients with SMM eventually do develop MM.26
the body; (2) detectable M-protein in the blood or urine; and (3) end organ damage resulting from the plasmacytosis.24 The end organ damage is often summarized by the acronym CRAB which stands for hypercalcemia, renal dysfunction, anemia, and bone lytic lesions. A recent update has been made to the definition of MM by the International Myeloma Working Group (IMWG) to define MM as the presence of clonal plasmacytosis in the marrow or a plasmacytoma plus one or more of the following biomarkers of malignancy: at least 60% bone marrow plasmacytosis, free light chain (κ:λ or vice versa) ratio of at least 100, or more than one focal lesion on a MRI study.25 The presence or lack of M-protein further defines the MM into secretory and nonsecretory types. Thus, a person may have a biomarker of malignancy without symptoms and still have the diagnosis of MM. The diagnosis of smoldering myeloma (SMM) is made when a person has a clonal marrow plasmacytosis and/or M-protein present yet does not otherwise fulfill other criteria for either MGUS or MM.26 Patients with SMM have an elevated risk of transformation to active MM at approximately 10% per year for the first 5 years from diagnosis, then 3% per year for the next 5 years, followed thereafter at a rate of 1% per year. Most patients with SMM eventually do develop MM.26
Like MGUS, MM is more common in older individuals, with a median age of 70. Twice as many men are affected as women. There is a racial disposition as well, with approximately twice the incidence in persons of African descent than that found in Caucasians.27 Exposure to pesticides imparts a slight risk in developing MM, as well as does chronic antigen stimulation from infection or autoimmune disease; however, most cases are idiopathic.28,29 Familial myeloma and monoclonal gammopathy have also been observed but occur relatively infrequently, at only two to four times the usual risk of developing MM, which is currently estimated at 6.5 per 100,000 persons per year.22,30
Clinical features of MM relate to the CRAB end organ damage and immunodeficiency.23 The most common presenting symptom of MM is a complication of a lytic bone lesion, present in approximately 60% of patients at diagnosis, often leading to pathologic compression fractures of the vertebral bodies. These bony lesions can be seen on x-ray, although the more sensitive techniques of MRI and PET-CT modalities are used with increasing frequency for imaging.31 Malignant plasma cells carve out niches in the marrow through manipulation of cytokines that limit osteoblast activity and stimulate increased osteoclastogenesis.32, This activity can lead to the development of lytic bone lesions which have a “punched out” appearance on x-ray. Osteoporosis is also a consequence of this heightened osteoclastic activity and contributes to compression fractures and bone pain which are among the most common presenting features of MM. The cytokine activity of the malignant plasma cell can lead to systemic hypercalcemia with symptoms of constipation, confusion, and ultimately renal failure.
Anemia is the most common presenting laboratory feature in MM and results in fatigue and reduced exercise tolerance. MM can lead to anemia through multiple mechanisms, including occupation of space in the marrow, renal complications, and systemic inflammation with high interleukin-6 levels, leading to increased hepcidin production characteristic of the anemia of chronic disease. The replacement of normal marrow elements with MM cells can occasionally lead to leukoerythroblastosis on a blood smear. More commonly, the M-protein interacts with sialic acid on the erythrocytes and promotes red blood cell stacking, giving rise to rouleaux. The presence of rouleaux will also increase the erythrocyte sedimentation rate. Renal failure occurs at presentation in about 20% of newly diagnosed patients usually due to cast nephropathy. Cast nephropathy occurs when excess light chains overwhelm the reabsorptive capacity of the proximal renal tubules, which leads to intratubal aggregation of light chains, tubular obstruction, and local inflammation with acute kidney injury. Hypercalcemia—with symptoms of constipation, abdominal pain, and confusion—is present at diagnosis less commonly in about 15% of patients and contributes to worsening of renal function.
The dysregulated immunoglobulin production in MM leads to a relative humoral immunodeficiency from immunoparesis (reciprocal reduction in uninvolved immunoglobulins). Twenty percent of patients with MM present with infection, most commonly from encapsulated bacteria such as Streptococcus pneumoniae and Haemophilus influenzae. Deposition of M-protein or free light chains on nerve endings can lead to peripheral neuropathy, present in about 15% of persons at diagnosis.33,34 Approximately 20% of patients are asymptomatic at diagnosis and are identified through screening laboratory testing alone, most commonly identified by an elevated serum total protein or increased globulin gap (serum total protein minus serum albumin).
MM is often defined by the isotype of M-protein secreted. IgG subtype is most common at 60% of cases, followed by IgA (20%), free light chain only (10%-15%), and IgD, IgE, IgM, and nonsecretory at approximately 1% each. Rarely, unchecked or undiagnosed MM can lead to the production of so much M-protein that hyperviscosity of the blood develops, leading to symptoms of blurry vision, headache, dyspnea, and bleeding. Hyperviscosity occurs most often in IgM lymphoplasmacytic lymphoma (WM) and occasionally in IgA myeloma, given that IgA and IgM are larger proteins (dimer and pentamer, respectively) and thus meet a lower threshold for interference with blood flow.9 Rarely it may be seen in patients with very high IgG levels and the rare myeloma patient with an IgM isotype. Hyperviscosity is treated initially with plasmapheresis; however, this is a temporary measure only and treatment must be directed at the underlying MM to prevent recurrence. Particular M-protein isotypes (often IgM) can also lead to type I cryoglobulinemia.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


