Abstract
The neurohormonal control of reproduction involves a network of central and peripheral signals in the hypothalamic-pituitary-gonadal axis. The gonadotropin-releasing hormone (GnRH) system plays a crucial part in this axis.
Integration of multiple signals ultimately results in neuronal release of GnRH, which then travels via the hypothalamic–hypophyseal portal circulation to the anterior pituitary gland, where it binds to the GnRH receptor 1 and upregulates synthesis and secretion of two pituitary hormones, luteinizing hormone (LH) and follicle-stimulating hormone (FSH). GnRH secretion is pulsatile with discrete bursts of GnRH affecting the release of pulses of LH and FSH by the pituitary. This chapter describes medical conditions as well as endogenous and exogenous factors that result in abnormalities of this complex hypothalamic-pituitary axis.
Keywords
Neurohormonal control of reproductive axis, physiology, pituitary
Reproductive dysfunction of central etiology can result from any process that disturbs the tightly regulated hypothalamic-pituitary system (see Chapter 1 ). The reproductive axis itself relies on pulsatile release of gonadotropin-releasing hormone (GnRH) from a collection of neurons that are distributed diffusely throughout the hypothalamus rather than within a discrete nucleus. Control of GnRH secretion is complex, including the kisspeptin ligand-receptor system described in detail as follows. Neuropeptide Y (NPY) facilitates GnRH release and gonadotropin responsiveness to GnRH. Substance P, beta-endorphin, leuenkephalin, and other endogenous opiates inhibit hypothalamic GnRH release, both acutely and tonically (in the case of beta-endorphin). Stress-related surges in corticotropin-releasing hormone (CRH) suppress GnRH gene transcription and GnRH release, as do inflammatory cytokines, such as tumor necrosis factor-α (TNF-α). This CRH-induced suppression may be mediated by endogenous opiates. The effects of catecholamines (norepinephrine, epinephrine, dopamine) are more controversial and likely depend on the prevailing endocrine milieu and the stage of sexual maturation. Neurotransmitters, such as gamma-aminobutyric acid (GABA), have been shown to have both excitatory and inhibitory effects in animal studies, and use of the GABA-ergic drug valproic acid (VPA) in women did not affect hypothalamic-pituitary hormones. Such conflicting data on the role of GABA in modulating GnRH release illustrate the challenges inherent in using rodent models to understand GnRH regulation in humans.
GnRH secretion is pulsatile, with discrete bursts of GnRH affecting the release of pulses of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) by the pituitary. Moreover, the frequency of GnRH secretion has been shown in animal models to differentially affect the synthesis of LH and FSH, with rapid-frequency GnRH pulses (1 pulse/hour) favoring LH synthesis and slow-frequency GnRH pulses favoring the synthesis of FSH. The secretion of LH and FSH is also regulated by direct negative feedback from gonadal products (estradiol [E 2 ] and inhibin A and B). Once released from the pituitary, LH and FSH travel through the systemic circulation to stimulate sex steroid production and gametogenesis by the gonads. The patterns of hypothalamic, pituitary, and gonadal activity are responsible for the transition from childhood to adulthood, as well as for normal ovulatory cycles in postmenarchal women and normal sexual function in males.
The presence of appropriate quantities of these hormones is not enough to maintain a normal reproductive axis. Frequent sampling of plasma hormone concentrations has led to the belief that most hypothalamic hormones are secreted episodically and often in a circadian rhythm. Hormonal release may be linked primarily to the sleep–wake cycle, synchronized with food ingestion, or related to the dark–light cycle. Thus integrity of the hypothalamic suprachiasmatic nucleus, which maintains both the circadian rhythm and the sleep–wake cycle, is an important factor for regulating gonadal function in men and women.
Patients with abnormalities of this complex hypothalamic–pituitary axis often show undetectable or inappropriately low levels of pituitary gonadotropins (FSH and LH) in combination with low sex steroid levels (testosterone <100 ng/dL in males and E 2 <20 pg/mL in females) (see Chapter 12 ). This combination of laboratory findings defines the condition hypogonadotropic hypogonadism. In contrast, patients with primary gonadal pathology have elevated FSH and LH levels in combination with low testosterone or E 2 levels, a disorder referred to as hypergonadotropic hypogonadism. The clinical presentation of hypogonadotropic hypogonadism varies with age of onset and degree of hormonal deficiency. Males may present with micropenis and cryptorchidism. Testicular function can be completely impaired with Leydig cell atrophy, or testicular size can approach normal with only mild Leydig cell impairment. The phenotype of hypogonadotropic hypogonadism in women is also quite variable, ranging from a classic eunuchoid appearance in some women (lower body segment [floor to pubis ] >2 cm longer than the upper body segment [ pubis to crown] and arm span >2 cm longer than height) to moderate breast development in others. Amenorrhea, whether primary or secondary, frequently is the reason why women seek medical attention. The degree of ovarian follicular development depends on the duration of gonadotropin deficiency; in cases of congenital hypogonadotropic hypogonadism (CHH), few follicles beyond the primordial stage may be present. The administration of exogenous gonadotropins or exogenous pulsatile GnRH can effect ovulation and pregnancy.
Although some patients with hypogonadotropic hypogonadism have no evidence of pulsatile LH secretion, others have detectable LH secretory pulses that are of insufficient frequency and amplitude to trigger gonadal steroid synthesis. A small number of patients show nearly normal patterns of LH secretion, but the LH molecules are inactive and consist only of the uncoupled α subunit. This heterogeneity of basal LH secretion in patients with hypogonadotropic hypogonadism is mirrored in their responses to exogenous GnRH stimulation. Complete unresponsiveness to GnRH is rare, but many patients show subnormal increases of FSH and LH. In other cases, FSH or LH levels may rise in isolation, or both may increase normally.
Hypogonadotropic hypogonadism may be physiologic, as occurs during puberty and lactation, or it may be a manifestation of congenital or acquired pathology. CHH usually occurs in the absence of other pituitary hormonal deficits, whereas acquired GnRH deficiency may be caused by tumors, infiltrative diseases, infection, trauma, or radiation, all of which can also cause other hormonal deficiencies. Epilepsy is frequently associated with reproductive dysfunction, such as the polycystic ovary syndrome (PCOS). Abnormalities of other hormones (e.g., thyroid hormone, cortisol, prolactin [PRL]), overnutrition and undernutrition, exercise, stress, and medications can also cause functional hypogonadotropic hypogonadism. Hypogonadotropic hypogonadism commonly occurs with pituitary disease due to tumors and apoplexy, as well as other autoimmune and inflammatory disorders.
Hypothalamic Dysfunction
- ◆
While there is an increasing knowledge about the genetic causes for both anosmic and normosmic idiopathic hypogonadotropic hypogonadism (IHH), the majority of cases of GnRH deficiency in humans are sporadic.
- ◆
Weight loss of 10% to 15% of normal weight is enough to delay onset of menarche and disrupts normal menstrual cycles in postmenarchal girls.
- ◆
A variety of medications can interfere with the hypothalamic-pituitary axis as a result of their tendency to increase prolactin. The degree of prolactin elevation is usually less than 100 ng/mL.
Reproductive integrity requires an intact hypothalamus, which secretes GnRH from neurons that are distributed diffusely throughout the hypothalamus rather than in a discrete nucleus. As discussed earlier, GnRH is secreted in a pulsatile fashion, the frequency of which differentially affects the pulsatile secretion of the gonadotropins. Moreover, the frequency of GnRH secretion is known to affect gene expression, with rapid-frequency GnRH pulses (1 pulse/hour) favoring LH synthesis and slow-frequency GnRH pulses favoring FSH synthesis. Pituitary LH and FSH secretion are also regulated by direct negative feedback from gonadal products (E 2 , inhibin A and B). The patterns of hypothalamic, pituitary, and gonadal activity are responsible for the transition from childhood to adulthood, as well as for normal ovulatory cycles in postmenarchal females and sexual function and spermatogenesis in males. Disruption of hypothalamic GnRH secretion at any point during development can have transient or permanent effects on fertility. Such disturbances can result from congenital, structural, or functional abnormalities of the central nervous system (CNS)-hypothalamic component of the reproductive axis.
Congenital Disease of the Hypothalamus
Hypogonadotropic hypogonadism resulting from abnormal development of GnRH neurons is relatively rare, affecting 1 : 10,000 males and 1 : 50,000 females. GnRH neurons originate during embryonic life outside of the CNS in the nasal placode. Olfactory neurons have a similar embryonic origin in humans and many other vertebrate species. Migration of olfactory axons toward the olfactory bulb (OB) anlage is required for normal OB development. GnRH neurons then migrate across the cribriform plate toward the OB in two phases, one before OB formation and one in association with OB formation. The latter wave of migration occurs in association with olfactory, vomeronasal, and terminal nerves as well as neural cell adhesion molecules, ultimately resulting in GnRH neuronal cell dispersion in the arcuate nucleus of the medial basal hypothalamus by 14 weeks of gestation. The mature GnRH axonal neuron network eventually coordinates pulsatile secretion of GnRH into the hypothalamic–hypophyseal portal circulation. Disruption of this complex series of events can result in varying degrees of hypothalamic dysfunction, which manifests clinically as hypogonadotropic hypogonadism.
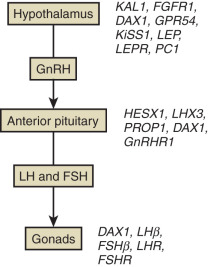
CHH is usually divided into two categories: that which is accompanied by olfactory deficits (anosmic IHH or Kallmann syndrome [KS]) and that which is associated with an intact sense of smell (normosmic IHH). Exciting progress has been made in identifying genetic causes for both anosmic and normosmic IHH, as summarized in Fig. 20.1 and Table 20.1 . Some genes are necessary for the correct embryonic differentiation of the GnRH-secreting neurons (fibroblast growth factor receptor 1 [FGFR1], fibroblast growth factor 8 [FGF8], negative elongation factor [NELF], heparin sulfate 6-O-sulfotransferase 1 [HS6ST1]) , chromodomain helicase DNA-binding protein 7 (CHD7), and sex determining region Y-Box 10 (SOX10). Others are essential for the migration of the GnRH neurons to the hypothalamus (KAL1[ANOS1], Prokineticin 2/Prokineticin 2 receptor 2 [PROK2/PROKR2], [CHD7]) ,WD repeat domain 11 (encoded by WDR11 ), semaphoring 3A (SEMA3A), and family zinc finger, and others are involved in the upstream signals which participate in the activation of the GnRH neuron (tachykinin 3/tachykinin 3 receptor [TAC3/TACR3], Kisspeptin 1/Kisspeptin 1 receptor [KISS1/KISS1R]) . The candidate genes also include GnRH, GnRH receptor, and the gonadotropins. The genetic basis is unknown for 60% of the normosmic hypogonadotropic hypogonadism. However, most cases of GnRH deficiency in humans are sporadic. In the cohort of 106 patients with GnRH deficiency with and without anosmia reported by Waldstreicher and colleagues, only 30% of cases were familial. Because IHH predominantly impairs fertility, families with multiple affected members are few, but the availability of gonadotropin therapy to induce fertility in both affected males and females may increase vertical transmission of the condition.
| Clinical Features | Condition | Gene | Localization | Inheritance | Site of GnRH Impairment |
|---|---|---|---|---|---|
| HH + anosmia | Kallmann syndrome | KAL1 | Xp22.3 | X-linked | Hypothalamus (incomplete GnRH neuronal migration) |
| HH ± anosmia ± midline facial defects | Kallmann syndrome | KAL2 | 8p11.2 | Autosomal dominant | |
| HH + anosmia | Kallmann syndrome | Autosomal recessive | |||
| HH | GnRH-R | 4q21.2 | Autosomal recessive | Pituitary (↓ GnRH-R activity) | |
| HH | GPR54 | 19p13 | Autosomal recessive | Hypothalamus | |
| HH + adrenal insufficiency | Adrenal hypoplasia congenital | AHC | Xp21 | X-linked | Hypothalamus Pituitary |
| HH | LH β | 19q13.3 | Autosomal recessive | Pituitary | |
| HH | FSH β | 11p13 | Autosomal recessive | ||
| Optic atrophy + midline CNS abnormalities + HH | Septooptic dysplasia | HESX1 | 3q21.1-21.2 | Autosomal recessive | Pituitary |
| Obesity + HH | Obesity syndrome | Leptin | 7q31.3 | Autosomal recessive | Hypothalamus |
| Obesity + HH | Obesity syndrome | Leptin receptor | 1q31 | Autosomal recessive | |
| Combined hormone deficiencies (short stature, hypothyroidism, HH) | PROP1 | 5q | Autosomal recessive | Pituitary |
Anosmic Idiopathic Hypogonadotropic Hypogonadism
KAL1 (ANOS1) Mutation
The pathologic association between IHH and olfactory dysfunction was originally recognized in the 19th century, but Kallmann and Schoenfeld first suggested a genetic basis for this group of patients in 1944. In 1989, Schwanzel-Fukuda and colleagues discovered a tangle of GnRH neurons on the surface of the cribriform plate in a human fetus with X-linked KS. The finding of the neurons in this location rather than in the typical location in the hypothalamus established defective GnRH neuronal migration as the cause of hypogonadotropic hypogonadism in KS. Because the initial differentiation and migration were normal, research then focused on factors affecting axonal growth, path-finding, and maturation. The KAL1 gene on the X chromosome (Xp22.3) was subsequently identified in 1991 by two groups, and both deletions and point mutations have since been found in patients with KS. The product of KAL1, anosmin-1, is transiently expressed in numerous tissues of the developing human fetus, including the OB and along the migratory pathway for GnRH neurons. In the absence of anosmin-1 (with the KAL1 mutation), OB differentiation and early olfactory axon navigation are impaired. Without olfactory nerve synaptogenesis in the OB, GnRH neurons have no migratory pathway to follow to the forebrain, resulting in the anosmia and hypogonadotropic hypogonadism typical of KS. KAL1 expression also occurs in the mesonephric tubules, ureteric bud, and corticospinal tract by 7 weeks of age and in the retina and kidney by 11 weeks of age. This tissue-specific expression accounts for the high prevalence of renal, neurologic, and midline facial abnormalities in patients with KS. Unilateral renal agenesis is believed to occur because of developmental failure of the collecting duct system in the absence of mesonephric tubule anosmin-1. Synkinesia (“mirror movements,” or the involuntary movements of one extremity when an individual is asked to perform rapid, repetitive motions with the other extremity) results from abnormal fast–conducting ipsilateral corticospinal tract projections. Absence of anosmin-1 expression in the facial mesenchyme leads to a high incidence of cleft palate in patients with KAL1 mutations. Renal agenesis and synkinesia were present in 31% and 85% of patients, respectively, of a British cohort of patients with KS. These abnormalities were not present in any patients with KS without the KAL1 mutation or in patients with normosmic IHH.
As is the case with all forms of hypogonadotropic hypogonadism, KS is much more common in males with prevalence estimated at 1 : 7500 males versus 1 : 70,000 females. The KAL1 mutation accounts for only 3% to 15% of cases of anosmic IHH and no more than one third of familial cases of IHH, but it is highly penetrant. No cases of KAL1 mutations have been described in female patients or in normosmic males. Patients with KS usually do not undergo puberty, but a minority may have some degree of testicular growth or breast development, suggesting that GnRH deficiency is not always complete. Additional reproductive features may include cryptorchidism and microphallus. As is the case with other genetic defects discussed later, there is no clear genotype-to-phenotype relationship in those with a KAL1 mutation, illustrating the importance of modifier genes and environmental factors.
KAL2 Mutation
Mutations in the gene encoding FGFR1, a tyrosine kinase receptor, also referred to as KAL2, appear to be inherited in an autosomal dominant pattern and are seen in 7% to 10% of individuals with KS. FGF signaling is involved with the formation, growth, and shaping of a variety of tissues and organs, including the OB. FGFR1 has been detected in both the nasal placode and the developing OBs of mice as well as along the migratory pathway for GnRH neurons and in the mature hypothalamus. Studies in rodents with targeted abolition of FGFR1 signaling resulted in aplasia of the OB. Both FGFR1 and anosmin-1 are coexpressed in the olfactory placode of human embryos by 4.5 weeks’ gestation and again at 8 weeks in the terminal nerve portion that guides GnRH neuronal migration to the hypothalamus. Studies suggest that FGFR1 is a ligand for anosmin-1. Loss-of-function mutations in the FGFR1 gene located on chromosome 8p11.2 can cause IHH with or without anosmia. At least 12 missense mutations have been identified in this gene thus far, and loss of function of the isoform FGFR1c is present in approximately 10% of male and female patients with KS. Isoform FGFR1c is also important for palate morphogenesis and olfactory development.
Unlike KAL1 mutations, KAL2 mutations are not always associated with a severe reproductive phenotype. A minority of patients may have delayed puberty or normal reproductive function, suggesting that the KAL2 mutation may not prevent GnRH migration entirely but may affect neuronal maturation in the hypothalamus. This variable expressivity of KAL2 suggests a role for other genes or the environment in modifying the phenotypic expression of this mutation. It also raises the possibility that KAL2 mutations account for other reproductive phenotypes associated with hypothalamic dysfunction, such as hypothalamic amenorrhea and delayed puberty.
CHH is less severe in patients with KAL2 mutations than in those with KAL1 mutations, and mean LH levels are significantly lower in KAL1 patients compared with KAL2 patients. Identification of KAL2 mutations and the relationship between FGF signaling and anosmin-1 may explain the male predominance of hypogonadotropic hypogonadism despite the low prevalence of the X-linked form of the disease. Females have two copies of the KAL1 gene and therefore higher levels of anosmin-1, which may compensate for inadequate FGFR1 function in the presence of a KAL2 mutation. Thus women with KAL2 mutations often have a milder, if not a normal, reproductive phenotype. On the other hand, males have a lower level of KAL1 or anosmin-1 expression and are unable to compensate for impaired FGF signaling in the presence of a KAL2 mutation. Thus their reproductive phenotypes tend to be more severe.
Other Candidate Genes
As is the case for IHH as a whole, most cases of KS are sporadic and cannot be traced to familial transmission. Autosomal recessive transmission of KS does exist, but the responsible gene has yet to be cloned. Multiple other candidate genes that may be involved in cases of anosmic IHH without KAL1 mutations have been identified in the past decade. Mutations of CHD7, which can cause CHARGE syndrome (coloboma, congenital heart disease, choanal atresia, mental and growth retardation, genital hypoplasia, and ear malformations or deafness), have been seen in KS patients. Heterozygous mutations in NELF have been reported in a few patients with KS, and the murine form of NELF is involved in olfactory and GnRH neuronal migration. Mutations in Pkr2, which encodes a G-protein-coupled receptor critical for OB and reproductive organ development in mice, have been found, as have mutations in the HS6ST1 gene. The enzyme is involved in the modulation of extracellular matrix components, such as heparin sulfate polysaccharides, and thought to modulate cell-cell communication, which is required for neural migration. Heterozygous mutations of WDR11 have been found in a small number of patients with KS. The WDR11 gene encodes a protein which interacts with the transcription factor EMX1. The interaction between both factors is important for the development of olfactory neurons. FGF receptor mutations have also been identified in normosmic individuals with IHH, suggesting that anosmic IHH and normosmic IHH are part of a spectrum of disease rather than distinct clinical entities.
Normosmic Idiopathic Hypogonadotropic Hypogonadism
Not all patients with hypogonadotropic hypogonadism have an impaired sense of smell. Those with a normal sense of smell are given the diagnosis of IHH. In the American cohort reported by Waldstreicher and colleagues, approximately half of patients with hypogonadotropic hypogonadism were normosmic. Nasal mucosal biopsy specimens from patients with normosmic IHH showed immunologically recognizable GnRH, providing evidence that the GnRH gene itself was intact and that defective GnRH synthesis was not the cause of IHH in these patients.
Gonadotropin-Releasing Hormone Mutations
There are also patients with IHH manifesting with severe hypogonadism who do have isolated GnRH deficiency. It was not until 2009 that a homozygous frameshift mutation in the GnRH1 gene in such patients was reported. GnRH1 encodes the preprohormone of GnRH. These patients had severe hypogonadism, suggesting that isolated GnRH deficiency can be a rare cause of IHH. Other etiologies for IHH have since been identified in this group of patients in whom the frequency of cryptorchidism and nonreproductive phenotypes (synkinesia, palate abnormalities, hearing loss) is much less common than in anosmic IHH.
Gonadotropin-Releasing Hormone Receptor Mutations
Attempts to identify congenital defects that would explain normosmic IHH have led to the discovery of loss-of-function missense mutations in the gene encoding the GnRH receptor (GnRH-R) on chromosome 4q21.2. These mutations are transmitted in an autosomal recessive pattern. The GnRH receptor is a G-protein-coupled transmembrane receptor involved with signal transduction. Mutations in this protein were initially considered unlikely because patients with normosmic IHH showed increased FSH and LH secretion in response to exogenous GnRH stimulation. However, in the study by de Roux and colleagues, the index cases both had partial IHH and likely responded normally to GnRH stimulation because the exogenous dose was high enough to overcome their partial receptor defects. GnRH-R mutations are estimated to occur in 40% of autosomal recessive cases of normosmic IHH and in 10% to 15% of sporadic cases. Heterozygotes are unaffected.
Identical mutations can produce different clinical phenotypes, suggesting that gonadotroph function depends on other genes as well. The clinical presentation of patients with GnRH-R mutations can include microphallus, cryptorchidism, gynecomastia, and delayed puberty in males, and primary amenorrhea, incomplete thelarche, and delayed puberty in females. Phenotypes can also be milder, as in men with normal adult-sized testes as well as intact spermatogenesis but impaired LH-dependent Leydig cell testosterone secretion (“fertile eunuch syndrome”). It is now believed that GnRH secretion in these cases is sufficient to trigger enough Leydig cell testosterone production locally to maintain spermatogenesis and testicular growth but insufficient to generate systemic testosterone levels capable of achieving full virilization.
Screening for GnRH-R mutations at the time of diagnosis of IHH is not clinically useful unless fertility is an immediate treatment goal. In these cases, knowledge of a GnRH-R mutation would guide one toward gonadotropin therapy and away from pulsatile GnRH therapy, as the latter was less effective in restoring reproductive function. As with other confirmed genetic abnormalities, identification of a GnRH-R mutation would also allow screening and treatment of family members.
GPR54 Mutations
Homozygous deletions in the gene GPR54 on chromosome 19 (19p13) were first reported in 2003 in a large consanguineous family with multiple members affected by IHH but with normal GnRH receptors. GPR54 encodes an orphan G-protein-coupled receptor that is expressed widely across the reproductive axis, including the hypothalamus, pituitary, gonads, and placenta. The ligand for GPR54 is kisspeptin (formerly metastin), which is derived from kisspeptin-1. Kisspeptin was originally discovered because of its ability to inhibit metastatic melanoma and breast cancer cell lines. Continued study of this protein suggests that it may play an important role in regulating central puberty through its ability to signal through GPR54 and stimulate GnRH release. GPR54 mutations account for 2% to 5% of normosmic IHH. Both male and female GPR54- deficient mice had a hypogonadotropic hypogonadal phenotype (males with small testes, females with delayed vaginal opening and delayed follicular maturation), but they had normal levels of hypothalamic GnRH and responded to exogenous GnRH. Similarly, spermatogenesis and successful ovulation have been achieved in GPR54- deficient patients treated with exogenous GnRH. GPR54 does appear to have tissue-specific effects, because the in vitro administration of kisspeptin metastin did not have the same stimulatory effects on pituitary gonadotrophs as it did on the hypothalamus, and homozygous mutations in GPR54 did not affect spermatogenesis or ovarian steroidogenesis. The exact mechanism by which GPR54 regulates puberty remains the subject of ongoing investigation.
Tachykinin 3 Mutations
Homozygous mutations in TAC3 , which encodes neurokinin B and its heptahelical transmembrane G-protein-coupled receptor TACR3 , have been described in patients with normosmic IHH. Patients with this mutation have very low basal LH secretion with a nonpulsatile pattern, while pulsatile GnRH treatment normalizes LH release.
Gonadotropin Mutations
Both FSH and LH are composed of a common α subunit and a β subunit that is unique to FSH, LH, thyroid-stimulating hormone (TSH), and human chorionic gonadotropin (hCG). Alpha subunit mutations have not been reported and likely are not compatible with life. However, beta subunit mutations in both the LH and FSH molecules have been identified and are associated with delayed puberty.
Pituitary Transcription Factor Mutations
Mutations in various homeobox transcription factors involved with normal adenohypophyseal development have been reported. Homozygous mutations in LHX3 have been described in two consanguineous families who presented with combined pituitary hormone deficiencies, including deficits in LH and FSH. LHX3 encodes a protein that is required for pituitary development and motor neuron specification. Mutations in PROP1 are also associated with variable degrees of FSH and LH insufficiency and other pituitary hormone deficiencies. PROP1 has both DNA-binding and transcriptional activation ability. Its expression leads to ontogenesis of homozygous and heterozygous mutations of HESX1 and has been implicated in some cases of septo-optic dysplasia, a disease that includes optic nerve hypoplasia, pituitary gland hypoplasia, and midline CNS anomalies. Children with septo-optic dysplasia can have either precocious puberty or pubertal failure.
A novel mutation in HESX1 has been described that causes combined pituitary deficiency but no septo-optic dysplasia phenotypes.
Hypogonadotropic Hypogonadism and Adrenal Failure
Normosmic hypogonadotropic hypogonadism can occur in association with adrenal failure due to loss-of-function mutations in DAX1 (adrenal hypoplasia congenital). DAX1 encodes an orphan nuclear receptor on chromosome Xp21 and is critical for the development and function of both the adrenal gland and the hypothalamic-pituitary-gonadal axis. In the presence of DAX1 mutations, adrenal failure usually presents in male infants or boys and is followed by impaired pubertal development due to failure of hypothalamic and pituitary gonadotropin production. Males with this mutation are often resistant to gonadotropin stimulation because of impaired spermatogenesis. Steroidogenic factor 1 (SF1), another nuclear receptor that regulates gene transcription in both the adrenal gland and the gonads, caused both adrenal and gonadal agenesis when knocked out in a murine model. Human SF1 mutations were associated with XY sex reversal and adrenal failure. It is now thought that DAX1 represses transcription of nuclear receptors, including SF1, and that loss of DAX1 function causes a variety of adrenal, pituitary, and hypothalamic abnormalities.
Hypogonadotropic Hypogonadism and Obesity Syndromes
Mutations in leptin and the leptin receptor have been identified in morbidly obese patients with normosmic IHH. It is leptin’s effects on the release of NPY that are likely responsible for its effects on GnRH secretion. Mutations in prohormone convertase 1 (PC1) also cause morbid obesity and hypogonadotropic hypogonadism, as well as hypocortisolemia (with elevated proopiomelanocortin) and hypoinsulinemia (with elevated proinsulin).
Adult-Onset Idiopathic Hypogonadotropic Hypogonadism
An acquired form of GnRH deficiency was described in 1997 in 10 men who presented with decreased libido, impotence, and infertility at a mean age of 35 years after undergoing normal puberty. None had features of congenital GnRH deficiency (anosmia, synkinesia, cleft palate, or a family history of GnRH deficiency), and none had a tumor, a history of radiation, infection, or infiltrative disease that might have explained their abnormal gonadotropin and sex steroid levels. Treatment with pulsatile GnRH reversed the hypogonadism and restored fertility in the subset of men who received long-term therapy. Recent data show that some men with IHH carry a mutation in the FGF8 gene and have normal reproductive function before onset of hypogonadism, suggesting a combination of genetic and environmental influences.
Diagnosis of Idiopathic Hypogonadotropic Hypogonadism
Hypogonadotropic hypogonadism should be included in the differential diagnosis for any patient with absent or incomplete pubertal development. In males, this can include microphallus, cryptorchidism, absence of facial hair, and small testes; and in women, this can include incomplete or absent breast development, amenorrhea, or a eunuchoid body habitus. Obtaining a family history can also be helpful when considering a diagnosis of IHH. Whereas the incidence of delayed puberty in the general population is less than 1%, the incidence of delayed puberty among relatives of a series of 106 patients with IHH was 12%. Because the GnRH pulse generator is quiescent until the onset of puberty (after an initial period of activation during infancy), the diagnosis of hypogonadotropic hypogonadism is usually not made until the age of 18 years, when such physical findings might prompt laboratory evaluation. Characteristic findings include low sex steroid levels (testosterone <100 ng/dL; E 2 <20 pg/mL) in association with low or inappropriately “normal” gonadotropin levels. Assessment of other pituitary hormones should also be performed when hypogonadotropic hypogonadism is initially discovered. Other biomarkers for the diagnosis of hypogonadotropic hypogonadism also include inhibin, INSL3, AMH, and kisspeptin. To exclude structural causes of GnRH deficiency, magnetic resonance imaging (MRI) is indicated in the evaluation of such patients. When laboratory studies and negative findings on imaging studies offer no other explanation for hypogonadotropic hypogonadism, a diagnosis of IHH or isolated GnRH deficiency is appropriate.
Sarfati et al. reported for the first time two cases due to KAL 1 and FGFR1 mutations in which the diagnosis was identified during fetal life on the basis of KS in the parents and noninvasive monitoring (fetal ultrasound).
Management of Idiopathic Hypogonadotropic Hypogonadism
Therapy for patients with IHH depends on the degree of sexual maturity and the desire for fertility. For men with severe gonadotropin deficiency who have yet to complete puberty, hCG is used as a surrogate for LH to stimulate Leydig cell testosterone secretion, typically using an intramuscular dose of 2000 IU 3 times weekly. Plasma testosterone and sperm counts are followed every 4 to 8 weeks, and hCG doses are adjusted to maintain normal testosterone levels. Recombinant FSH (75 to 150 IU 3 times weekly) is often required to improve the quality of the sperm produced, although it may not be required for initiation of spermatogenesis in men with partial IHH and those with postpubertal onset of hypogonadism. Inability to achieve a normal sperm count after 6 months of hCG treatment alone and declining sperm count despite hCG are indications to add FSH. Success with this combination is possible, regardless of previous exposure to testosterone, although individuals differ in the timing of their response to induction of spermatogenesis. Restoration of fertility occurs as early as 4 months in some men, but it can take more than 1 year in others. Even if spontaneous pregnancy does not occur, adequate sperm are usually present after combined hCG and FSH therapy such that assisted reproductive techniques, including in vitro fertilization with intracytoplasmic sperm insertion, are good options. Subcutaneous or intravenous administration of GnRH via pump has been shown to achieve spermatogenesis more rapidly than treatment with gonadotropins. GnRH is not currently available in the United States but is available in Austria, Canada, France, Germany, Hong Kong, Italy, South Korea, The Netherlands, Singapore, Sweden, and Switzerland. Once pregnancy is achieved, hCG but not FSH should be continued to maintain normal serum testosterone and some spermatogenesis to facilitate future pregnancies. Men who only desire virilization can be given intramuscular or topical testosterone depending on their preferred mode of administration.
Cyclic estrogen and progesterone should be given to female patients to induce normal sexual development and preserve normal bone mineral density (BMD), although progesterone should not be added until breast development is complete. Women who desire fertility should receive exogenous gonadotropins to stimulate ovulation. Recombinant FSH alone can induce follicular growth, as shown in Fig. 20.2A , and has been shown to be as effective as highly purified FSH in achieving pregnancy with a lower risk of ovarian hyperstimulation. However, women with severe hypogonadotropic hypogonadism require some source of LH to generate adequate androgen precursors for aromatization to E 2 , as illustrated in Fig. 20.2B . E 2 is required for endometrial proliferation and creation of a uterine environment optimal for implantation.
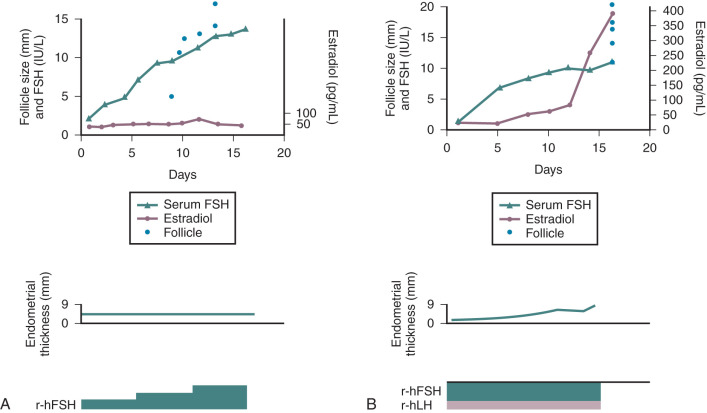
Treatment with hCG (75 IU daily) may be used in such cases to maintain adequate E 2 concentrations. In women with baseline LH levels of greater than 1.2 IU/L, FSH alone may be sufficient to stimulate follicular growth as well as ovarian steroidogenesis. Once adequate follicular development and E 2 -induced endometrial proliferation have occurred, exogenous recombinant hCG (250 µg) can be used to induce ovulation and is as effective as urinary hCG. Recombinant formulations of LH are available, but their shorter half-life and higher cost preclude their widespread clinical use. Provision of luteal-phase progesterone is also recommended. When using FSH and hCG in combination, a woman’s chance of conception is approximately 70% after six cycles. Such treatment does carry a risk of ovarian hyperstimulation and multiple gestations, and these risks are higher than those associated with pulsatile GnRH therapy. For this reason, close monitoring with transvaginal ultrasound is required in women receiving gonadotropin therapy. In addition, pregnancies in women with hypopituitarism should be considered high risk because they are associated with higher rates of spontaneous abortion, possibly reflecting a uterine environment devoid of important endocrine growth factors.
A report by Chan et al. described the reversal of GnRH release in a male patient after infusion of kisspeptin. In addition to issues surrounding hormone replacement therapy and fertility treatment, there are other considerations when treating patients with IHH. Those with anosmic IHH and a family history suggestive of an X-linked mode of inheritance should have an abdominal-pelvic ultrasound at the time of diagnosis to assess for possible renal agenesis, given the relatively high frequency of this nonreproductive abnormality in patients with a KAL1 mutation. Genetic testing is not routinely employed in patients with IHH, because the majority of cases are sporadic. However, limited genetic testing is available for patients suspected of carrying a KAL1 mutation. Research laboratories can also investigate for point mutations in KAL1 or FGFR1 and GnRHR gene mutations.
Prognosis for Recovery
Until recently, it was believed that lifelong hormone therapy was required to maintain secondary sex characteristics and sexual function in men with IHH, but recent reports of testicular growth in men receiving maintenance androgen replacement therapy led to the identification of a minority of patients (10%) in whom reversal of IHH occurs. Based on the retrospective study by Sidhoum et al., lifetime incidence of spontaneous recovery might be as high as 22%. In addition, an increased number of neurokinin B mutations have been found in reversible cases. Such reversal can occur in men who underwent partial puberty or who were prepubertal at the time of diagnosis and in men with either anosmic or normosmic IHH. It is believed that exposure to sex steroids promotes plasticity of the GnRH neuronal network such that gradual reversal of IHH is possible, even in the face of genetic defects in men who are treated with androgen replacement therapy. Thus men being treated for IHH should have their hypothalamic–pituitary axis reassessed periodically so that such reversal can be identified.
Structural Disease of the Hypothalamus
Mass lesions in the hypothalamus can also disrupt the secretion of GnRH into the hypothalamic-hypophyseal portal circulation and cause hypogonadotropic hypogonadism. Such cases usually also cause other pituitary hormone abnormalities. Distinguishing hypothalamic mass lesions from pituitary mass lesions requires MRI. Dynamic testing with hypothalamic-releasing hormones is not used clinically, with the exception of CRH, which is used in combination with inferior petrosal sinus sampling (IPSS) to determine whether hypercortisolemia is of pituitary or ectopic origin.
Tumors
Craniopharyngioma
Craniopharyngiomas are rare, almost always benign, epithelial tumors arising from remnants of the craniopharyngeal duct that occur with an annual incidence of 0.13 cases per 100,000 person-years. They are the most common lesions involving the hypothalamic-pituitary region in children and account for 5.6% to 15% of the intracranial neoplasms in this population (2% to 5% in adults). Craniopharyngiomas affect men and women at equal rates, and no genetic susceptibility has been identified. They are histologically benign lesions that are usually cystic or mixed (84% to 99%) rather than solid (1% to 16%). Two subtypes have been identified: adamantinomatous and papillary. The adamantinomatous type is more common in children and often contains calcification. Although tumor margins are usually sharp, adamantinomatous craniopharyngiomas tend to generate significant reactive gliosis in the adjacent normal brain tissue, making complete surgical resection difficult. Cysts may be multiloculated and contain viscous fluid rich in desquamated squamous epithelial cells and cholesterol. Because of its sometimes brown-green appearance of this cystic fluid, the term “motor oil” has been used to describe it. A recent preliminary study has shown that nuclear accumulation of beta-catenin is present in adamantinomatous craniopharyngiomas but not in papillary craniopharyngiomas. The strong beta-catenin expression indicates the reactivation of the Wnt signaling pathway, which is involved in the development of several neoplasms. The papillary subtype is seen almost exclusively in adults and is rarely associated with calcification. Papillary craniopharyngiomas tend to be solid or mixed rather than purely cystic, and they are less likely to infiltrate surrounding normal brain tissue, making surgical resection easier. Papillary craniopharyngiomas usually lack the peripheral palisading of epithelial cells, cystic change, calcification, and the cholesterol depots found in the adamantinomatous type.
Recent genetic analysis has shown differences between these two subtypes of craniopharyngiomas in 60% to 96% of adamantinomatous craniopharyngiomas showing mutations in B-catenin (CTNNB1) , which is a downstream effector of the Wnt pathway that is involved in growth and development.
Papillary craniopharyngiomas have been discovered to frequently have a mutation of the BRAF gene, which plays a role in the mitogen-activated kinase pathway.
Craniopharyngiomas almost always have a suprasellar component and are rarely purely intrasellar, as shown in Fig. 20.3 . Because of their proximity to vital neural structures and their significant size at the time of diagnosis (58% to 76% are between 2 and 4 cm), they frequently present with headache, nausea, vomiting, visual disturbances, growth failure (in children), and hypogonadism (in adults), although the rate of tumor growth also determines the severity of symptoms. Bitemporal hemianopsia is the most common visual complaint, occurring in almost 50% of cases. Hydrocephalus can be present, occurring more frequently in children than in adults for unclear reasons (41% to 54% vs. 12% to 30%). With the exception of hydrocephalus and its associated symptoms, there was no difference in symptom duration or extent of hormone deficiency between children and adults with craniopharyngiomas.
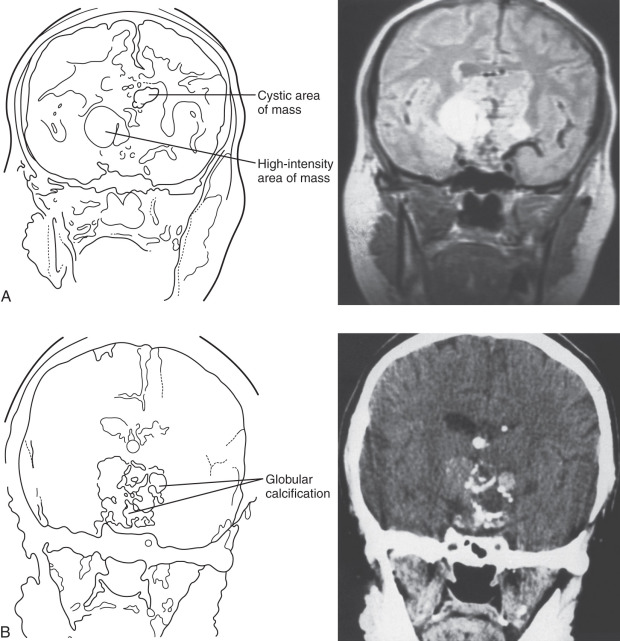
Both computed tomography (CT) and MRI can be helpful in diagnosing craniopharyngiomas. CT can show calcification that is characteristic of the adamantinomatous subtype, whereas MRI with gadolinium enhancement provides better structural analysis. The appearance of the craniopharyngioma depends on the proportion of solid and cystic components and the content of the cyst. The solid tumor part is iso- or hypointense compared with the brain on precontrast T1 sequences and shows enhancement after contrast. On T2 sequences, the signal is mixed, hypo-, or hyperintense. The cystic component is hypointense on T1 and hyperintense on T2. If there is protein, cholesterol, or methemoglobin present, then there may be a high T1 and a low T2 signal. The cyst can show a peripheral contrast-enhancing rim in the T1 postcontrast images. When the findings of imaging studies are consistent with craniopharyngioma, surgical resection is usually the initial therapy, although radiation therapy (RT) has been used for small lesions not causing pressure-related visual, neurologic, or endocrinologic damage. The surgical approach depends on tumor size, location, and degree of calcification as well as the experience of the surgeon, but it often involves craniotomy and is sometimes a two-stage process. Preoperative drainage of large cystic components in the days before surgery can relieve pressure-related symptoms and make surgical resection easier, although cyst cavities refill if surgery is delayed by weeks. Gross total resection is the goal in all cases, but it often cannot be achieved because of tumor size and adherence to vital neurovascular structures.
Unfortunately, craniopharyngiomas often recur even after gross total resection has been achieved and confirmed radiographically. Recurrence rates at 10 years vary from 0% to 62% but improve with use of adjuvant RT. Recurrence-free survival correlates best with extent of surgical removal; age, sex, location (intrasellar vs. suprasellar), and size of the tumor at the time of surgical resection do not predict recurrence. Some reports suggest that recurrent craniopharyngiomas show a higher microvessel density, lower levels of galectin-3 and macrophage migration inhibiting factor, and lower levels of retinoic acid receptor beta and higher levels of retinoic acid receptor gamma. Management of recurrent disease usually involves RT rather than repeated surgery because of the perioperative morbidity and mortality associated with the latter. In fact, surgery for recurrent disease is only recommended in the event of acute pressure-related symptoms. The 10-year progression-free survival rate from the time of the first recurrence was 72% in a series of 25 patients who received RT. Other options for the management of recurrences include brachytherapy (for predominantly cystic lesions), bleomycin installation via an Ommaya reservoir, and stereotactic radiosurgery.
Some studies have demonstrated some response after systematic chemotherapy in pediatric patients after treatment with interferon (INF)-α-2b (pegylated and nonpegylated) and offer hope for future therapies with lower side effect profiles.
The endocrine, visual, and neuropsychologic morbidities associated with craniopharyngiomas are significant. At the time of presentation, 35% of the patients in Van Effenterre’s cohort had one to three hormone deficits. After treatment, the frequency of individual hormone deficits varies significantly across studies (88% to 100% for growth hormone [GH], 80% to 95% for FSH and LH, 55% to 88% for adrenocorticotropic hormone [ACTH], 39% to 95% for TSH, and 25% to 86% for antidiuretic hormone [ADH]) and is not affected by the type of therapy. Unlike pituitary adenomas, it is very unusual for patients with pretreatment hormone deficits to experience restoration of those axes after treatment, but pituitary hormones can be replaced relatively easily. Visual defects (i.e., quadrantanopia or worse) are present in the majority of patients after surgery with or without adjuvant RT. Potentially more disabling are the consequences of hypothalamic damage that occur so commonly in patients with craniopharyngiomas. Obesity secondary to hyperphagia affects 26% to 61% of patients who undergo surgery with and without adjuvant RT. Data by Bomer et al. suggest that development of obesity in craniopharyngioma patients is associated with lower resting energy expenditure compared with obesity patients without craniopharyngioma, suggesting that factors independent of hyperphagia might play a role. Diabetes insipidus (DI), with an impaired sense of thirst resulting in water and electrolyte imbalances, was present in 14% of a pediatric cohort. Neuropsychologic and cognitive functions often decline in patients with craniopharyngiomas. At follow-up 7 years after initial treatment, 16% of the adults and 26% of the children in Van Effenterre’s cohort were not living independently and had not returned to their previous jobs or schools. Mortality rates 3 to 6 times higher than those of the general population have been reported in patients with craniopharyngiomas.
Rathke Cleft Cyst
RCCs are the most common incidentally discovered sellar lesion. They are found in 13% to 33% of normal pituitary glands. They are discovered usually later in life with a peak between the 4th and 6th decades, which is most likely because they grow slowly. and symptoms occur later in life. RCC originates from embryonic remnants of the Rathke pouch, which extends cranially to form the craniopharyngeal duct. The pouch is derived from the ectodermal stomadeum (primitive oral cavity). The Rathke pouch cells proliferate and form the pars anterior of the pituitary and the cells from the posterior wall differentiate into the pars intermedia. The pouch involutes and the craniopharyngeal duct obliterates. If the obliteration of the lumen does not occur, then a cyst develops and the remnants in the craniopharyngeal canal develop into cystic lesions ranging from simple RCCs to complex craniopharyngiomas. RCCs are benign lesions that can range from a few millimeters to 5 cm. The content of the cyst is mostly thick and can consist of cholesterol and proteins. The cyst wall is lined by cuboid or columnar epithelium and occasionally with goblet cells secreting mucous into the cyst. RCCs and craniopharyngiomas have overlapping pathologic features and are sometimes difficult to distinguish. Coexistence of RCCs with pituitary adenomas has been shown in 0.5% to 1.7%. Of these adenomas, GH secreting pituitary adenomas and prolactinomas are the most frequently described coexisting with RCCs. Location of RCCs are usually in the pars intermedia sometimes with suprasellar extension. Symptoms usually do not occur until the cyst is large enough to put pressure on the surrounding structures. The most common symptom includes headaches. The presence of headaches does not correlate with the cyst size but seems to be related to either high or isointense content on T1-weighed MRI, mucous cyst content, or cystic wall inflammation. Major visual field defects are present in up to 75% of cases, and anterior pituitary hormone deficits are present in 19% to 81% of patients. Hypogonadism because of hyperprolactinemia is the most frequent hormonal manifestation. MRI often shows a hypointense lesion with rim enhancement and a hyperintense signal in the T-2 weighted images. Hyperintensity on T1-weighted images and isointensity on T2-weighted images suggest mucoid material is present in the cyst; hyperintensity in both T1- and T2-weighted images suggests the presence of blood. Cysts with high protein content show a high T1 signal intensity. Only 13% of RCCs show calcification on CT. Asymptomatic, small RCCs are treated conservatively without surgery. If symptomatic, the aim is to drain the cyst content and to remove as much of the capsule as possible. Radiation is usually not used routinely for treatment with the exception of recurrent cysts. The 4-year recurrence rate has been reported to be 48%; the presence of squamous metaplasia in the cyst wall is associated with a higher risk of recurrence.
A recent update of the Cochrane review comes to the conclusion that there are currently no well-powered randomized controlled trials to give recommendations for the use of bleomycin in the treatment of cystic craniopharyngiomas in children.
Germ Cell Tumors
Germ cell tumors (GCTs) are believed to result from malignant transformation and abnormal migration of primordial germ cells. They are characterized by their secretion of hCG and alpha-fetoprotein (AFP). Intracranial GCTs occur most commonly in the pineal region (50%) and in the anterior hypothalamus (30%). They account for 2.9% of all intracranial tumors in children and have a peak incidence between ages 10 and 14 years old. Presenting symptoms depend on tumor location: nausea, vomiting, diplopia, and paralysis of upward gaze (Parinaud syndrome) occur with pineal gland tumors, whereas suprasellar lesions can cause hypopituitarism, DI, and visual deficits. Histologically, GCTs are often cystic and are classified as germinomatous GCT (GGCT) or nongerminomatous GCT (NGGCT). NGGCTs account for the minority of intracranial GCTs and include choriocarcinomas, teratomas, embryonal carcinomas, and yolk sac tumors. Serum hCG and AFP levels tend to be elevated in NGGCTs, whereas only a minority of patients with GGCTs have an abnormal serum hCG. Stereotactic biopsy is indicated when the diagnosis is unclear from imaging studies and serum hCG and AFP levels. If a diagnosis of GGCT is made, therapy usually consists of radiation rather than resection because of the radiosensitivity of GGCTs and the neurologic morbidity associated with surgery. Chemotherapy is an alternative therapy, but it is associated with a higher recurrence rate when used in the absence of RT. NGGCTs are less radiosensitive and, therefore, carry a poorer prognosis. However, platinum-based chemotherapy, in combination with craniospinal RT, is often used and followed by surgical resection if a residual mass persists.
Other lesions occurring in the pineal region include pineal parenchymal tumors, astrocytomas, meningiomas, metastases, cysts, and vascular malformations. Hypothalamic-pituitary function can be disturbed by any of these.
Infiltrating Diseases
Sarcoidosis
Sarcoidosis is a multisystem disease of activated T lymphocytes and noncaseating granulomas that commonly affects the lungs, lymph nodes, eyes, and skin, but can involve the CNS and peripheral nervous system in 10% to 20% of cases. Neurologic symptoms can be the presenting sign of disease in 50% of the latter group of patients and can increase morbidity and mortality. CNS lesions can appear as subdural plaques or infundibular plaques, infiltrating intraparenchymal pseudotumoral masses, or as multiple nodules on MRI studies. Although hypothalamic-pituitary lesions may not be evident on MRI, 25% to 33% of patients with neurosarcoidosis have DI, and autopsy studies in such patients show extensive granulomatous inflammation of the hypothalamus with little pituitary involvement. DI results from vasopressin deficiency and destruction of neurons in the neurohypophysis, but damage to the osmoreceptor controlling thirst also contributes to polyuria and polydipsia. Other symptoms of hypothalamic disease include somnolence, body temperature dysregulation, hyperphagia, and obesity. Impaired secretion of anterior pituitary hormones also occurs; GH and gonadotropin deficiencies occur more commonly than TSH or ACTH deficiency. In Murialdo and Tamagno’s Italian series of 91 patients, 39% had hypogonadotropic hypogonadism and 59% of the women in the study ( n = 46) reported amenorrhea.
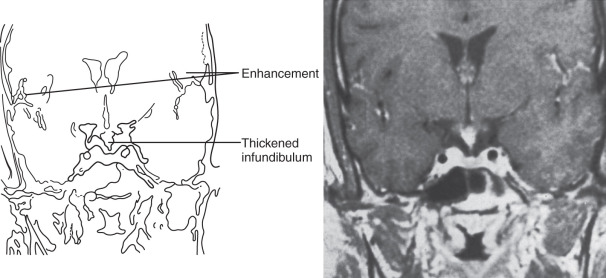
As with other forms of sarcoidosis, confirmation of neurosarcoidosis requires histologic evidence of noncaseating granulomas and lymphocyte infiltration of affected tissue, but obtaining nervous system tissue can be difficult. However, a probable diagnosis can be made with the proper clinical picture and imaging studies (i.e., MRI; Fig. 20.4 ). PRL elevation can be a useful screen for hypothalamic involvement. Results of cerebrospinal fluid (CSF) analysis are abnormal in 80% of patients with CNS sarcoid and can include elevations in protein, lymphocytes, or angiotensin-converting enzyme levels.
Prednisone (up to 40 to 80 mg daily) is the mainstay of treatment for neurosarcoidosis, as it is for other forms of the disease, although therapy is often intensified in CNS disease. Cyclosporine A (4 to 6 mg/kg) is often added to minimize glucocorticoid requirements. RT is employed in patients who do not respond to oral medications. Unfortunately, anterior pituitary deficiencies usually are not restored despite treatment. Dopaminergic agents can be used to decrease PRL levels, but they will not restore fertility if hypogonadotropic hypogonadism is also present.
Sarcoidosis can also infiltrate the epididymis and testes, resulting in painless epididymitis or a scrotal mass, both of which respond to glucocorticoids. Granulomatous involvement of the uterus and fallopian tubes has also been reported in women with sarcoidosis who present with amenorrhea or menorrhagia. Thus reproductive dysfunction in patients with sarcoidosis can be both primary and secondary to CNS disease.
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is another multisystem disease of unknown etiology that has a predilection for the hypothalamus and pituitary. It is characterized by the clonal accumulation and proliferation of abnormal dendritic histiocytes with accompanying lymphocytes, eosinophils, and neutrophils. These cellular infiltrative lesions can destroy a variety of tissues, including skin, bone, lymph nodes, liver, spleen, lungs, and bone marrow. Known previously as Letterer-Siwe disease, Hand-Schüller-Christian disease, eosinophilic granuloma, and histiocytosis X, LCH primarily affects children between the ages of 1 and 3 years old. It can remit spontaneously or disseminate and can even result in death in rare cases. Adults make up fewer than 30% of all reported cases of LCH with an incidence of 1.8 cases per million people. LCH in adults more commonly affects the skin, lung, and bones.
The hypothalamic-pituitary axis is involved in 5% to 50% of children with LCH, with 15% to 50% of these children presenting with DI. The incidence of DI increases over time. It is usually seen within 5 years of diagnosis and occurs more often in the setting of multisystem disease. DI was also the earliest hormonal abnormality in the adult cohort reported by Kaltsas and colleagues; 33% of the patients presented with DI. Anterior pituitary hormone deficiency occurs in approximately 20% to 35% of patients and is usually associated with DI. In a large pediatric cohort, the estimated 5- and 10-year risks of pituitary involvement with LCH were 22% and 24%, respectively. GH deficiency is the most common pituitary endocrinopathy, occurring in 10% of patients with LCH and increasing in frequency over time. Ear, nose, and throat involvement increases the risk of GH deficiency. GH deficiency in the absence of DI is unusual, but 54% of patients with DI who were followed for 10 years ultimately had GH deficiency. GH replacement effectively improves the final height in children with GH deficiency, although midparental height is usually not achieved because children have fairly severe growth restriction at the time therapy is initiated. Importantly, GH therapy did not increase the risk of LCH disease activity. GH replacement in adults with LCH has not been well studied.
Gonadotropin deficiency also occurs in LCH and is almost always in association with other anterior pituitary hormone deficiencies. Because most patients are prepubertal at the onset of disease activity, those with gonadotropin deficiency require exogenous sex steroids to induce and maintain puberty. Fifty percent of the cohort of 12 patients reported by Kaltsas and colleagues had gonadotropin deficiency at a median of 7 years old after diagnosis of LCH, and exogenous gonadotropins successfully restored fertility in 1 male patient.
Findings on MRI are abnormal in the majority of patients with LCH and endocrinopathy. Loss of the posterior pituitary bright spot on T1-weighted images and infundibular enlargement are the most common abnormalities, with the former observed in 100% of patients with DI. As shown in Fig. 20.5 , other less common radiologic abnormalities include a thickened infundibular stalk, a partially or completely empty sella, and lesions in the hypothalamus.
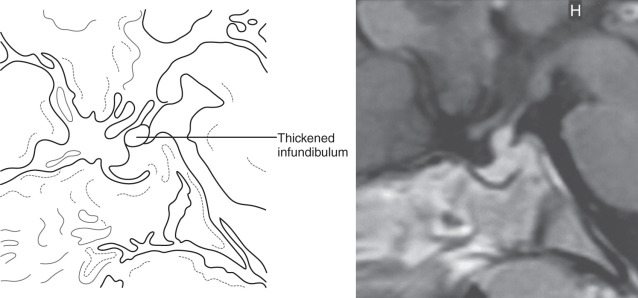
Both children and adults with LCH and DI are at high risk for anterior pituitary deficits and should be followed closely. Unfortunately, dynamic evaluation of pituitary function was not a useful predictor of later endocrinopathies. DI and other hormonal deficiencies tend to be permanent once established, despite treatment of LCH lesions with RT, chemotherapy, or both. However, RT may be helpful for controlling localized disease and mass effects. Chemotherapy was not helpful in long-term disease management in adults.
Other infiltrating diseases that can cause hypothalamic infiltration with resulting reproductive dysfunction include hemochromatosis (which more commonly affects the pituitary; discussed later in the chapter), leukemia, lymphoma, and Wegener granulomatosis. Central DI is often the presenting feature, and treatment of the underlying condition sometimes results in resolution of this and other associated hormonal abnormalities. When associated with malignancies (leukemia, lymphoma), hypothalamic involvement is a poor prognostic feature and usually does not respond to treatment.
Infections
Infections are a rare cause of hypothalamic–pituitary disease affecting the reproductive axis. They usually present acutely with fever, headache, meningism, and change in mental status or seizure, and can be identified using culture, polymerase chain reaction, or detection of antibodies to viral antigens. Tuberculosis and atypical mycobacterial infections, viral encephalitis, mycotic infections, and bacterial meningitis can all cause infiltration of the hypothalamus or pituitary and result in hormonal deficiencies. Box 20.1 summarizes the classification of infiltrative, infectious, and other nontumoral diseases involving the hypothalamus that can cause hypogonadotropic hypogonadism.
Infiltrative or Autoimmune Diseases
Sarcoidosis
Langerhans cell histiocytosis
Lymphomatous diseases
Castleman disease
Wegener granulomatosis
Vasculitis
Infectious Diseases
Tuberculosis
Atypical mycobacterial infections
Bacterial meningitis
Encephalitis (viral)
Mycotic infections
Others
Trauma
Pituitary stalk section
Radiation therapy
Hydrocephalus and pseudotumor cerebri
Head Trauma
Head trauma has been recognized as a cause of neuroendocrine dysfunction for many years, but assessment of hypothalamic–pituitary function during acute and long-term management of patients with traumatic brain injury (TBI) occurs infrequently. TBI is most common in males between the ages of 11 and 29 (5 : 1 male-to-female ratio) and is the leading cause of death and disability among young adults. Road accidents account for 75% of cases of TBI. Most patients who have post–head trauma hypopituitarism (PHTH) were comatose for a period after their injuries, and all had Glasgow Coma Scale scores of less than 13. PHTH can result from structural injury to the hypothalamus or pituitary, but a vascular insult leading to cerebral swelling is the most likely mechanism in most cases. Such localized edema increases intracranial pressure in the sellar or suprasellar regions and can temporarily impair hypothalamic–pituitary function. PHTH may also be an adaptive mechanism to the acute illness often associated with TBI.
One third of patients studied 3 months to 23 years after head injury had at least one pituitary hormonal deficiency: 18% had an inadequate GH response to insulin tolerance testing, and 20% to 25% had insufficient responses to GnRH stimulation. The frequency of gonadotropin deficiency was even higher (80% to 90%) when defined by low early morning testosterone or E 2 levels and inappropriately low FSH and LH levels. TBI-associated neuroendocrine dysfunction only presents with DI in the minority of cases (25% to 30%). PRL elevation occurs in approximately 50% of patients and correlates negatively with Glasgow Coma Scale score, likely because it is a marker of pituitary stalk or hypothalamic damage.
Unfortunately, PHTH is usually not diagnosed during hospitalization unless a patient has acute ACTH deficiency or DI. Because pituitary hormone deficiency eventually recovers in approximately 60% of patients with moderate to severe head injury, some deficiencies (i.e., GH, gonadotropin) may never be recognized. Gonadotropin secreting cells appear to be the most sensitive to TBI, and patients with PHTH often present after hospital discharge with amenorrhea, erectile dysfunction, and infertility. FSH and LH in such patients may be inappropriately low. Most hormonal deficits (75%) appear within 1 year after injury, but diagnosis can be delayed by 5 or more years in 15% of patients. In patients with a known history of TBI whose anterior pituitary function is initially normal, repeat evaluation during follow-up care may be indicated given the delayed appearance of hypopituitarism in some cases.
Radiation Therapy
Hypothalamic–pituitary dysfunction after cranial irradiation has long been recognized to occur in survivors of childhood cancer and in patients who receive high-dose RT for nasopharyngeal carcinoma or pituitary disease. However, not until recently has the high frequency of endocrine dysfunction in adult survivors of nonpituitary brain tumors who receive RT been recognized. This frequency is only likely to increase with the improvements in prognosis for both children and adults with primary brain malignancies. Of a cohort of 56 patients with primary brain tumors who had received their last dose of RT at least 1 year before the time of study, 23 (41%) showed evidence of hypopituitarism with multiple (25%) or single (16%) hormone deficiencies. GH deficiency was most common, followed by gonadotropin, ACTH, and TSH deficiency. DI is rarely associated with radiation-induced hypopituitarism. The risk of hormonal deficiency depends on the total dose, fraction size, number of fractions received, and total duration of RT. Panhypopituitarism can occur when the total dose exceeds 60 gray (Gy), but this risk can be reduced by administering more fractions of smaller dose, thereby extending treatment time.
The rates of gonadotropin and GH deficiency in adults are significantly lower than those observed in pediatric studies, in which 20% to 50% of patients eventually have partial or severe gonadotropin deficiency, and GH deficiency occurs in 80% to 100% of patients. It is thought that the hypothalamic–pituitary axis is more radiosensitive in children than in adults. Doses exceeding 50 Gy in childhood can cause gonadotropin deficiency, but smaller doses can result in precocious puberty. In low-dose cranial irradiation used prophylactically in children being treated for acute lymphocytic leukemia, girls are more susceptible to early puberty than boys, but this sexual dichotomy disappears when higher doses (25 to 50 Gy) are used to treat primary brain tumors. In a pediatric cohort with preexisting GH deficiency, the relationship between age at time of irradiation and age at onset of puberty was linear. The severity of gonadotropin deficiency varies in postpubertal subjects from low normal to frankly low sex hormone levels.
The site of radiation-induced injury is the hypothalamus, and postradiation studies show normal responses to hypothalamic-releasing hormones, including GnRH. The mechanism of this radiation-induced damage is unclear but may involve direct damage to hormonally active cells or to their surrounding microvasculature. Alternatively, radiation may damage the vascular connections between the hypothalamus and the pituitary. Hormone deficiencies may not become evident for many years, because the radiation-induced damage takes its toll on the slowly dividing cells of the hypothalamus and secondary pituitary atrophy occurs after previous hypothalamic damage. The delayed appearance of these deficits mandates that such patients be followed annually for these late effects of radiation.
Gonadal damage is also a concern in patients treated for primary brain tumors, but this usually occurs as a result of adjuvant chemotherapy or spinal RT. Alkylating agents, procarbazine, cisplatin, and vinblastine are chemotherapeutic agents often associated with gonadal toxicity, whereas gonadal damage secondary to spinal RT occurs from radiation scatter. Sixty-four percent of girls who received craniospinal irradiation but no chemotherapy had evidence of primary ovarian failure over 8 years of follow-up. Sex steroids and germ cells are both lost in female patients treated with RT or chemotherapy, whereas males may present with infertility with normal levels of testosterone as a result of a differential effect of cancer therapy on Leydig cell function and spermatogenesis.
Treatment for radiation-associated infertility secondary to disruption of the hypothalamus or pituitary requires replacement of gonadotropins. Physiologic exogenous pulsatile GnRH therapy restores ovulation in the majority of women. Theoretically, cryopreservation of embryo, sperm, oocyte, or ovarian tissue can be used to preserve fertility when cancer treatment is expected to cause gonadal damage. However, each has significant drawbacks and none is routinely employed.
Seizure Disorders
Epilepsy is a relatively common condition, with a prevalence of 5 : 1000 to 9 : 1000 and has been associated with subfertility in numerous epidemiologic studies. The background rate of menstrual dysfunction (50% to 60%) far exceeds that of the general adult female population. PCOS is the most common form of reproductive dysfunction in this population and affects up to 25% of epileptic women, although hypothalamic amenorrhea and luteal phase deficiency (LPD) can also occur. The diagnosis of PCOS was unrelated to the type of epilepsy.
Both seizures themselves and the antiepileptic drugs (AEDs) used to treat them have been implicated as the cause for reproductive dysfunction in patients with epilepsy. Epileptiform discharges transiently increase PRL and may disrupt GnRH pulsatility, a hypothesis suggested by observations of altered LH pulse frequency in regularly menstruating women with epilepsy who were not taking AEDs. The similar rates of PCOS among medicated and unmedicated women with epilepsy also provide evidence that seizure activity itself interferes with normal reproductive function. In addition, the use of AEDs (specifically, VPA) in normal women and those with other conditions (bipolar affective disorder) does not increase the risk of PCOS or affect baseline LH pulse dynamics, gonadal hormones, or rates of oligomenorrhea, arguing for the position that epilepsy itself is responsible for interfering with reproductive function in some women.
AED therapy, especially VPA, appears to worsen the central reproductive dysfunction caused by epilepsy itself. VPA has been shown to augment LH secretion in response to GnRH in healthy women, presumably via its stimulatory effects on glutamic acid decarboxylase and its inhibitory effects on GABA transaminase, ultimately leading to an increase in brain GABA activity. No association between PCOS and the use of any specific AED was apparent in a relatively lean cohort of epileptic women reported by Bilo and colleagues, but polycystic ovaries and hyperandrogenism were observed in 70% of a Finnish cohort receiving VPA monotherapy. The frequency of menstrual disorders, polycystic ovaries, and hyperandrogenism was increased in both lean and obese women but was higher in the latter. Anovulatory cycles occurred in 43% of women who were currently using VPA or had done so within the previous 3 years, whereas only 10% of cycles were anovulatory in women with generalized epilepsy who had never taken VPA. Although VPA is associated with weight gain, the changes observed in serum androgens (testosterone, androstenedione) occurred as early as 1 to 3 months after initiation of VPA and before any significant changes in body weight. These observations suggest that VPA-associated PCOS in women with epilepsy is likely multifactorial in origin, with genetic and environmental factors playing some role. In addition, the initiation of VPA before the age of 20 years was a risk factor for future development of PCOS. Although VPA has never been shown to increase LH secretion, it may have direct ovarian effects, which, in combination with the GnRH pulse generator dysfunction described earlier, ultimately lead to the hyperandrogenemia characteristic of many epileptic women receiving VPA.
Ovulatory dysfunction related to AEDs may also arise as a result of alterations in cytochrome P450 activity. Phenytoin, phenobarbital, and carbamazepine induce cytochrome P450 activity, thus increasing sex hormone–binding globulin (SHBG) synthesis and reducing bioavailable testosterone and E 2 levels. VPA inhibits cytochrome P450 enzyme activity and increases androgen concentrations. However, these changes in bioavailable hormone levels are not always significant enough to alter reproductive function, and women taking carbamazepine have never been shown to have a higher risk of menstrual dysfunction, hyperandrogenemia, or PCOS.
Unfortunately, anovulatory cycles can be the only sign of epilepsy-associated reproductive dysfunction. Thus an awareness of the effect of the disease and its treatment on patients’ reproductive health should be part of routine care for women with epilepsy. Menstrual cycle duration and body weight should be monitored regularly in women taking AEDs, especially VPA, so that reproductive abnormalities are not overlooked. When such changes develop, switching women to other AEDs, such as lamotrigine, in consultation with their neurologist, can reverse the adverse reproductive and metabolical phenotype associated with AED treatment.
Men with epilepsy also have reproductive endocrine abnormalities and are 36% less likely than unaffected male siblings to be biological fathers. Levetiracem has been shown to have an impact on basal, but not gonadotropin-stimulated testosterone and estrogen secretion when tested in in vitro studies. In clinical studies, no changes on testosterone levels were found.
Approximately one-third of men with temporolimbic epilepsy have hypogonadism, and reductions in sperm count or motility as well as abnormal sperm morphology are observed in 8% to 90% of men regardless of AED use.
Functional Alterations and Disorders of the Hypothalamus
Physiologic Hypogonadotropic Hypogonadism
Pubertal Transition
Hypogonadotropic hypogonadism can be physiologic during certain phases of life. GnRH neurons are temporarily activated during infancy, producing midpubertal levels of either testosterone or E 2 (see Chapter 17 ). After 6 months of age, however, GnRH neurons become quiescent, and gonadotropin levels and sex steroid levels fall. This physiologic hypogonadotropic hypogonadism of prepuberty persists until approximately 8 to 9 years of age, when serum LH, FSH, and E 2 levels begin to rise, primarily overnight. Over the course of puberty, mean LH increases 116-fold, FSH increases 7-fold, and E 2 increases 12-fold. These changes are shown in Fig. 20.6 .
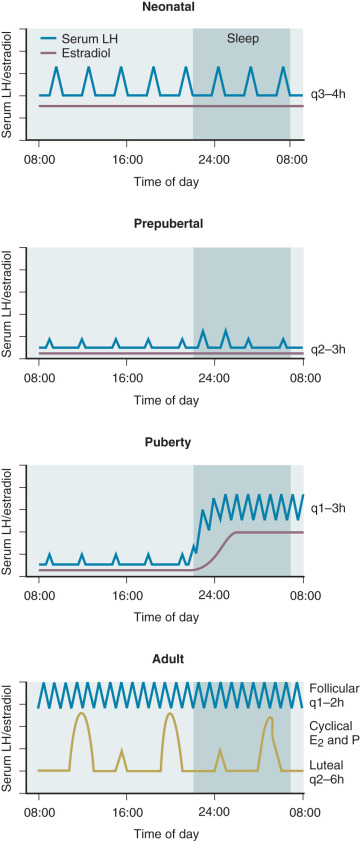
These gonadotropin changes directly reflect hypothalamic GnRH release because they can be simulated by exogenous GnRH administration, can be blocked by GnRH antagonist administration, and can occur in the absence of functioning gonadal tissue. The factors mediating this reactivation of the GnRH pulse generator in early puberty are still unclear, as are the factors permitting the progressive increase in daytime LH that occurs during puberty. Because hypogonadotropic hypogonadism is normal during puberty, distinguishing it from pathologic causes of hypogonadotropic hypogonadism becomes difficult during adolescence. Both are characterized by absent or incomplete sexual maturation, low gonadotropin concentrations, low sex steroid levels (testosterone <100 ng/dL; E 2 <20 pg/mL), and no evidence of other hypothalamic–pituitary axis abnormalities. The α subunit response to GnRH injection was used in the past to separate these two conditions; adolescents who ultimately had the former condition had little α subunit production after GnRH, indicating pituitary disease. However, where GnRH is unavailable, clinicians must rely on clinical features and family history rather than laboratory data to distinguish constitutional delay of growth and puberty (a normal variant characterized by hypogonadotropic hypogonadism) from more sinister causes of delayed puberty.
Postpartum Period
The postpartum period is another time in life in which hypogonadotropic hypogonadism is physiologic. Amenorrhea occurs in all women after childbirth. High levels of E 2 and progesterone during pregnancy are initially responsible for this hypothalamic-pituitary suppression. However, amenorrhea persists for varying durations into the puerperium and is prolonged further in women who breastfeed. FSH levels remain low (similar to those seen in the early follicular phase), mean LH and LH pulse frequency are reduced, and serum E 2 and progesterone are low. The mechanisms responsible for this continued suppression of the hypothalamic–pituitary–gonadal axis are unknown. PRL has been implicated because it is universally elevated in the immediate postpartum period and suppresses GnRH release. However, PRL declines long before menstrual cyclicity returns in lactating women, arguing that mechanisms other than hyperprolactinemia must be responsible for lactational amenorrhea. The suckling stimulus itself increases PRL, as shown in Fig. 20.7 , but also increases the sensitivity of the GnRH pulse generator to the negative feedback effects of E 2 , such that GnRH and LH pulse activity are nearly absent in the immediate postpartum period. However, both of these effects of suckling diminish with time.
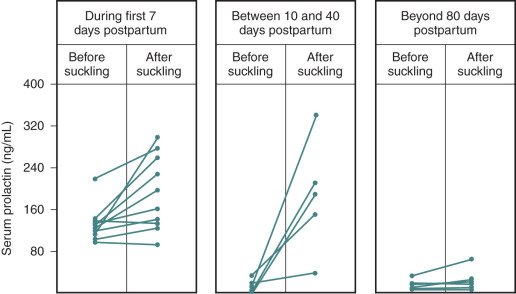
Once they reappear, LH pulse patterns are initially erratic and are augmented by sleep, similar to the pattern observed in early puberty. Follicle growth and associated E 2 production often return to normal before ovulation resumes, because suckling impairs the normal positive feedback of E 2 that is required for the preovulatory LH surge. Thus menses may return, but cycles may be anovulatory until the suckling stimulus decreases further and normal ovulatory cycles resume. Additional evidence suggesting a hypothalamic etiology for lactational amenorrhea comes from studies in lactating women exposed to pulsatile exogenous GnRH. Midcycle LH surges occurred in these women, luteal phases were normal, and FSH declined appropriately in response to increasing levels of ovarian steroids. The hypothalamic hypogonadism of lactation can persist for more than 12 months depending on the frequency and intensity of suckling. This endogenous contraception allows women to cope with the physiologic demands of the postpartum period before considering additional pregnancies. Both puberty and the postpartum period illustrate the neuroendocrine control mechanisms that govern GnRH, and abnormalities can be seen in a number of pathophysiologic states.
Pathophysiologic Hypogonadotropic Hypogonadism
Hypogonadotropic Hypogonadism Associated With Altered Secretion of Thyroid Hormone, Cortisol, and Prolactin
The neuroendocrine components of the reproductive axis are highly sensitive to disturbances in other hormones, including thyroid hormone, cortisol, and PRL. Within this context, menstrual irregularity occurs in approximately 25% of women with any degree of thyroid hormone deficiency, although the prevalence increases with the severity of hypothyroidism. Oligomenorrhea and menorrhagia are the most common menstrual disturbances, and sexual dysfunction (hypoactive sexual desire, erectile dysfunction, and delayed ejaculation) is observed in the majority of men with hypothyroidism. Men with hypothyroidism often have low free testosterone levels that improve with initiation of thyroid hormone replacement. Hypogonadotropic hypogonadism (low FSH and LH) may be present, but the clearance rate of the gonadotropins and sex steroids is reduced in the presence of hypothyroidism. Pituitary sensitivity to GnRH may be reduced in hypothyroid children, but it appears normal in hypothyroid women. Fewer than 10% of patients with newly diagnosed hypothyroidism (TSH > 4 mU/L) have associated hyperprolactinemia that resolves with initiation of thyroid hormone replacement. However, correction of hyperprolactinemia in these patients does not correct the menstrual dysfunction. Only with restoration of a euthyroid state does menstrual function normalize, assuming it was secondary to thyroid dysfunction originally.
Thyroid hormone excess, as occurs in Graves disease, is also associated with menstrual irregularities, anovulation, and infertility in women and is associated with sexual dysfunction in men. Amenorrhea may be present in severe hyperthyroidism. Excess thyroid hormone increases gonadotropin sensitivity to GnRH and may impair aromatase activity. Hepatic SHBG production is also increased in hyperthyroidism, resulting in higher total testosterone and E 2 , but normal free testosterone. This increase in E 2 can cause gynecomastia in men with severe hyperthyroidism. Spermatogenesis may be impaired in men with thyroid hormone excess but normalizes after restoration of a euthyroid state.
Reproductive dysfunction frequently accompanies abnormalities of cortisol secretion . Amenorrhea develops in 25% of patients with adrenocortical insufficiency, although the etiology of such a severe reproductive disturbance is likely multifactorial, with chronic illness, weight loss, or autoimmune ovarian failure contributing. Appropriately treated patients with autoimmune adrenal insufficiency (Addison disease) have basal LH levels and LH pulse patterns similar to those of normal healthy adults. During experimental periods with low cortisol replacement, patients with Addison disease showed elevated CRH and ACTH levels, but normal sex steroid levels (testosterone and E 2 ). Gonadotropin alterations were gender-specific; mean LH concentrations and LH pulse amplitudes were suppressed in men in the setting of insufficient cortisol replacement for 47 hours, whereas similar conditions had no effect on LH dynamics in premenopausal and postmenopausal women. These differences may reflect higher opioid receptor activation in men than in women because opioids mediate the inhibitory effects of glucocorticoids on LH release. Responses to low-dose GnRH stimulation did not vary in either sex under low cortisol conditions, although higher doses of GnRH did alter stimulated FSH and LH concentrations. These experimental conditions do not replicate the effects of long-term hypocortisolism, however, and clinical observations would suggest that maintaining physiologic cortisol levels is crucial for preserving a normal hypothalamic–pituitary–gonadal axis.
Menstrual irregularities (amenorrhea, oligomenorrhea, polymenorrhea) are present in the majority of women with cortisol excess (Cushing syndrome), with only 20% reported to have normal menses in one large cohort. GnRH neurons possess cortisol receptors, and hypercortisolemia blocks both GnRH release and the action of gonadotropins on the gonad. Gonadotropin levels are low or inappropriately normal in the presence of glucocorticoid excess, although GnRH testing showed normal or slightly increased gonadotropin reserve in hypercortisolism. Thus impaired GnRH release from the hypothalamus rather than pituitary dysfunction likely explains the reproductive pathology seen in Cushing syndrome. This reduction in gonadotropin secretion explains the radiographic appearance of the ovaries in women with Cushing syndrome, which tend to be smaller, with few primordial follicles and stromal hyperplasia. Plasma E 2 and free and total testosterone are low in hypercortisolism, which is consistent with hypogonadotropic hypogonadism. E 2 correlates inversely with the degree of cortisol elevation. This hypogonadism can be reversed by restoration of normal serum cortisol levels.
Although excess cortisol itself could explain these abnormalities of the hypothalamic–pituitary–gonadal axis, other hormones have been implicated, including corticotropin-releasing hormone, PRL, and androgens . CRH is an important inhibitor of gonadotropin release in settings of acute stress, but it is suppressed by negative feedback in states of cortisol excess and is therefore unlikely to play a role in menstrual dysfunction in patients with Cushing disease. Similarly, hyperprolactinemia is present in the minority of patients with Cushing disease but does not correlate with the severity of the reproductive disturbance and is unlikely to be causative. Androgens were long believed to mediate Cushing-associated menstrual abnormalities. However, in a cohort of 45 premenopausal women with newly diagnosed Cushing disease, serum testosterone and androstenedione concentrations were elevated in only 29% and 39%, respectively, and there was no correlation between androgen levels and cortisol. Metyrapone treatment of women with Cushing syndrome restores regular menses despite the increase in androgen levels associated with this drug. These data suggest that hypothalamic GnRH release is suppressed by excess cortisol itself rather than by its adverse effects on other hormonal parameters.
Hyperprolactinemia is a common cause of reproductive dysfunction in premenopasual women and is usually caused by pituitary lactotroph adenomas that are less than 1 cm in diameter (microadenomas) (see Chapter 3 ). Hyperprolactinemia accounts for 10% to 20% of cases of amenorrhea (after pregnancy is excluded). Menstrual disturbances of varying degrees were present in 87% of a cohort of premenopausal women with hyperprolactinemia, whereas galactorrhea, another manifestation of hyperprolactinemia, was only present in 47%. The severity of menstrual disturbance is related directly to the degree of hyperprolactinemia. Mild hyperprolactinemia (20 to 50 ng/mL) may impair progesterone secretion by the corpus luteum, resulting in a shortened luteal phase but with no menstrual irregularities. Moderate increases in PRL (50 to 100 ng/mL) can result in oligomenorrhea, whereas amenorrhea and overt hypogonadism occur when PRL is significantly elevated (>100 ng/mL). However, any degree of hyperprolactinemia can contribute to infertility. The severity of reproductive dysfunction in men also relates to the degree of hyperprolactinemia and can include decreased libido, infertility, gynecomastia, and galactorrhea. The diagnosis of hyperprolactinemia is often delayed in postmenopausal women because they are already hypoestrogenic; such patients often present with compressive symptoms related to PRL-secreting adenomas, including headache, visual symptoms, or abnormalities of other pituitary hormones.
The mechanism of PRL-associated amenorrhea is suppression of hypothalamic GnRH release, resulting in low or inappropriately normal gonadotropin levels and low sex steroid levels. This reduction in hypothalamic GnRH release results in decreased LH pulse frequency, but LH pulse amplitude is higher. The sleep-associated slowing in LH pulse frequency that occurs in normal adults is also lost in those with hyperprolactinemia. These effects of PRL on the hypothalamus are not mediated by increased hypothalamic opioid activity, and can be overcome with provision of exogenous pulsatile GnRH, which has resulted in pregnancy. These observations and others show that hyperprolactinemia, whether due to normal physiologic states (lactational amenorrhea) or pathophysiologic causes, does not interfere with pituitary-gonadal feedback mechanisms or with the actions of LH and FSH on the ovary. Animal data suggest a suppressive effect of prolactin on kisspeptin expression in the arcuate nucleus neurons as a pathway through which hyperprolactinemia might inhibit LH secretion. Restoration of normal PRL levels, either medically or surgically, allows LH pulse frequency and amplitude to recover, thereby normalizing menses, correcting hypogonadism, and restoring fertility.
Evaluation of any woman with menstrual dysfunction or any man with symptoms consistent with hypogonadism should include measurement of a serum PRL level. Mild increases in PRL can occur with intense breast or chest wall stimulation, strenuous exercise, and emotional or physical stress. Thus mild elevations should be confirmed before further evaluation for a cause of hyperprolactinemia. PRL secretion is normally inhibited by tonic hypothalamic dopamine secretion; thus use of any drug that inhibits hypothalamic release or transport of dopamine to the pituitary or that blocks pituitary dopamine receptors can cause hyperprolactinemia. Such agents include metoclopramide, phenothiazines, butyrophenones, risperidone, verapamil, and, rarely, the selective serotonin reuptake inhibitors. Discontinuation of potentially offending agents should be attempted, if possible, for at least 72 hours to determine whether hyperprolactinemia is drug-induced. In the absence of drug-induced hyperprolactinemia, hypothalamic lesions (craniopharyngiomas, granulomatous infiltration) must also be considered in the differential diagnosis. Such processes can cause hyperprolactinemia by compressing the pituitary stalk and thus interfering with dopamine transport. PRL levels greater than 200 ng/mL should also be repeated after dilution to avoid the “hook” effect, which can artificially lower actual PRL values by interfering with both immunoradiometric and chemiluminescent assays. Once the presence of non–drug-induced hyperprolactinemia is confirmed, pituitary imaging with MRI is preferred for identification of adenomas.
Effects of Nutrition on the Neuroendocrine Components of the Reproductive Axis
States of Undernutrition: Effects of Fasting
Nutritional status is an important regulator of reproductive health (see Chapter 18 ). Individual and species survival are at stake when food is scarce; thus reproductive potential is appropriately reduced in the setting of undernutrition. The effects of undernutrition on the reproductive axis have been studied experimentally by fasting normal subjects. These studies have suggested that the hypothalamic–pituitary axis in men may be more sensitive to the effects of starvation than that in women. Mean LH and FSH decrease within 48 hours of fasting in men. Although the mechanisms involved remain unclear, similar reductions observed in animal studies did not appear to be opioid-mediated. The data on LH pulse frequency and amplitude are less consistent, however. Within this context, Veldhuis and colleagues did not detect any change in LH pulse frequency in healthy men during a 5-day fast, although LH pulse amplitude was attenuated. In contrast, the results of Cameron and colleagues in healthy men who fasted for 48 hours showed a significant reduction in LH pulse frequency, but no change in pulse amplitude. Serum testosterone decreased in both studies. The GnRH pulse generator in men may be more sensitive to the effects of fasting because of the sensitizing effects of gonadal steroids. Older men, whose sex steroid levels have declined with age, do not show the same fasting-induced changes in LH pulsatility that are seen in younger men. Animal studies have also shown that these effects of sex steroids on neuroendocrine function may vary with the type of stressor.
The GnRH pulse generator in women appears less susceptible to the effects of acute fasting. Despite minor perturbations in LH pulse frequency and mean LH, at the end of a 72-hour follicular-phase fast, follicle development in fasting women was similar to that in women who were fed, as illustrated in Figs. 20.8 and 20.9 .
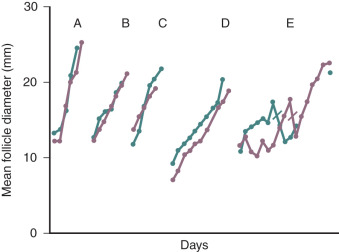
This resistance of the GnRH pulse generator to the acute effects of fasting occurs despite documented changes in cortisol and melatonin, both of which have been proposed to be critical regulators of the GnRH axis. Body weight may mediate the effect of fasting on GnRH drive, and LH pulse frequency declined by 20% in lean women compared with normal weight women. Attempts to determine the effects of acute undernutrition on GnRH activity during the luteal phase in women have proven more difficult, and exogenous administration of E 2 and progesterone to simulate the luteal phase also suppresses LH. As in their male counterparts, however, women showed alterations in both cortisol and melatonin when fasted during a simulated luteal phase, with dramatic increases in cortisol and duration of nocturnal melatonin secretion with fasting, evidence that this nutritional stress was not without some hormonal perturbation.
Fasting can only be maintained in healthy subjects for short periods. The effects of more sustained caloric deprivation have been assessed in healthy women maintained on 800 to 1100 kcal/day diets for one menstrual cycle. Such caloric restriction was sufficient to cause weight loss and resulted in anovulation in some women. Dieting reduced follicular-phase LH secretion as well as follicular-phase LH pulse frequency, but luteal-phase LH patterns and FSH were unaffected. Sex steroids (testosterone, androstenedione) were decreased, SHBG increased, and dehydroepiandrosterone sulfate did not change with dietary restriction. These data in healthy, regularly ovulating women suggest that cortisol and melatonin may be the first hormones affected by short-term fasting, but that continued caloric deprivation reduces GnRH activity within weeks. Amenorrhea can develop with loss of approximately 10% of body weight regardless of initial body weight.
States of Undernutrition: Anorexia Nervosa
Anorexia nervosa (AN) is an extreme example of the effects of undernutrition on the reproductive axis. It is difficult to separate the neuroendocrine effects of this nutritional deprivation itself from the hormonal alterations caused by the comorbid psychiatric illnesses that are frequently associated with AN. However, the effects of long-term caloric deprivation alone are also difficult to discern in other states of chronic undernutrition, such as protein–calorie malnutrition in hospitalized patients. Thus AN has been best studied as a model of the reproductive consequences of severe caloric restriction. AN is defined by weight loss of greater than 15% for height, an intense fear of gaining weight, a disturbance in one’s body image, and amenorrhea for at least 3 months. It is estimated to affect 0.5% of young women, with an even lower prevalence in males. Both primary and secondary amenorrhea can occur in AN, depending on the time of onset of the condition. A loss of 10% to 15% of normal weight for height is enough to delay the onset of menarche (primary amenorrhea), and a similar amount of weight loss disrupts normal menstrual cycles in postmenarchal girls (secondary amenorrhea). However, amenorrhea precedes weight loss in a significant percentage of patients with anorexia, evidence that psychologic stress and excessive exercise can also play a role. Other organ systems affected by AN include the cardiovascular and peripheral vascular (hypotension, arrhythmias), hematologic (anemia), dermatologic (lanugo hair), and renal systems (hypokalemia).
Menstrual disturbances in anorexia result from hypothalamic rather than pituitary or ovarian dysfunction. Mean serum LH concentrations are lower than those of regularly menstruating women studied in the follicular phase, and the LH secretory pattern is prepubertal, even in previously menstruating adolescents. Responses to exogenous GnRH are blunted; the degree of response is greater in women who are closer to ideal body weight and in those who will ultimately resume normal menses after restoration of body weight. Positive feedback from E 2 on the hypothalamus is also impaired in anorexic patients, as shown by studies with clomiphene citrate and exogenous estrogen that did not augment gonadotropin secretion. The reduction in gonadotropin stimulation of the ovaries results in low estrone (E 1 ), E 2 , progesterone, testosterone, and androstenedione levels. Reduction in aromatase activity as a result of low body fat also contributes to the hypoestrogenism seen in anorexia, as does the preferential synthesis of catecholestrogen over E 2 . Catecholestrogen is an endogenous antiestrogen that is capable of binding to estrogen receptors without having any biological action. Catecholestrogen also competes with dopamine for the enzyme catechol- O -methyltransferase, thus increasing dopamine levels, further inhibiting GnRH pulsatility, and potentiating hypoestrogenism. These alterations in E 2 metabolism result from changes in body weight and are not specific to anorexia. Additional evidence that anorexia-associated amenorrhea is the result of hypothalamic dysfunction is the induction of menses in anorexic women with the use of pulsatile GnRH.
The biochemical factors responsible for the hypothalamic amenorrhea in anorexia are unclear. Increased opioid tone is unlikely to be the only operative mechanism because administration of naloxone increased LH secretion in some, but not all, studies. Women who had reached ideal body weight and those with the highest basal LH levels had the most significant responses to opiate antagonists (naloxone). Dopamine can also inhibit LH release, but the use of the dopamine agonist metoclopramide did not result in a significant increase in LH in a cohort with anorexia. Metabolites of serotonin, such as 5-hydroxyindoleacetic acid, which stimulate hypothalamic GnRH release and increase pituitary sensitivity to GnRH, are low in patients with anorexia and increase during recovery, as shown in Fig. 20.10 .
Selective serotonin reuptake inhibitors, which restore CNS serotonin levels, have been shown to be effective in reversing abnormal eating behaviors in some patients with anorexia. Thus disturbance of the serotonin system may also play some role in anorexia-associated hypogonadotropic hypogonadism. Another potential factor is leptin, which is secreted by adipocytes and is critical for energy homeostasis. Leptin receptors have been identified in the hypothalamus, pituitary, endometrium, ovary (granulosa, theca, and interstitial cells), and testes (Leydig cells). Leptin indirectly stimulates GnRH release, acting in concert with other molecules involved in appetite control (NPY, melanocortin). It also directly stimulates LH and FSH release from anterior pituitary cells. Leptin gene expression is promoted by estrogen and insulin. Anorexia and other states of hypoinsulinemia are characterized by reduced leptin concentrations, which correlate with body fat. Weight restoration in anorexia is associated with increases in leptin, changes that correlate with increases in serum LH and FSH. Thus leptin may serve as a peripheral signal of adequate adipose stores that is capable of reactivating the hypothalamic–pituitary–gonadal axis and restoring menses during recovery from AN. The changes in leptin in AN are also accompanied by changes in ghrelin, another peptide intimately involved in control of feeding behavior. Ghrelin is increased in anorexic females compared with age-matched control subjects and constitutionally thin women without AN. Ghrelin correlates negatively with body mass index (BMI) and leptin, and partial weight recovery results in a 25% decrease in ghrelin levels in patients with anorexia.
Dehydroepiandrosterone sulfate and androstenedione are reduced in women with anorexia, but serum and urine cortisol levels are higher than normal. This preferential synthesis of cortisol over adrenal androgens is similar to that seen normally in prepubertal children and reflects relative 17,20-desmolase deficiency. The resulting hypercortisolemia is augmented by the prolonged plasma half-life of cortisol in AN. In spite of elevated cortisol, ACTH levels are not elevated at baseline or in response to CRH, as illustrated in Fig. 20.11 .
These findings show that the negative feedback effects of this hypercortisolemia remain intact at the level of the pituitary and that excess secretion of CRH and decreased clearance of CRH contribute to this hypercortisolemia. The lack of clinical signs of hypercortisolemia may result from diminished tissue expression of glucocorticoid receptors in patients with anorexia. Importantly, these alterations of the hypothalamic-pituitary-adrenal (HPA) axis resolve with long-term weight recovery. The hypothalamic-pituitary-gonadal axis also recovers with restoration of ideal body weight, as shown in Fig. 20.12 .
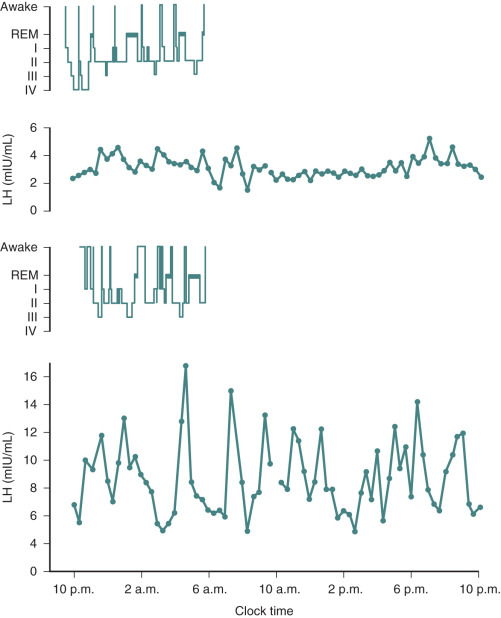
Although no absolute BMI or percentage of body fat has been found to correlate with the resumption of menses, more than 85% of patients who achieve a weight that is 90% of their ideal body weight note return of menses within 6 months. However, amenorrhea persists in a minority of women (10% to 30%), despite weight gain. Of the latter, many admit to restricting their fat intake and score higher on questionnaires designed to detect anorexic behaviors. Hypoestrogenemia in such women correlates with the degree of ovarian dysfunction, and ovarian ultrasound shows small, amorphous ovaries. However, small multifollicular ovarian cysts appear once weight gain starts, with the return of a dominant ovarian follicle once premorbid weight is achieved. Those who recover from anorexia, resume regular menses, and have normal eating behaviors can successfully conceive and bear healthy children.
Recovery from AN and restoration of ideal body weight requires both emotional and nutritional rehabilitation, and remains quite challenging despite improvements that have been made in understanding the pathophysiology of AN during the last century. Standardized mortality rates in patients with AN are increased 10-fold to 13-fold when patients are followed for 10 years. The crude death rate in large meta-analyses, including more than 3000 patients with AN, was 5.9%. The percentage of patients who recover entirely is less than 50%, whereas a significant proportion of patients have binge-eating behaviors at some point during their illness. At least 10% still meet the criteria for AN after long-term follow-up. Factors associated with poorer outcomes included longer duration of illness before the first inpatient treatment and a lower BMI, illustrating the importance of early diagnosis and intervention.
Unfortunately, the effects of severe undernutrition on other body systems can persist even when anorexic patients restore their body weight. Osteopenia and osteoporosis are the most serious long-term consequences of AN, and they result from both decreased bone formation (related to energy deprivation) and increased bone resorption (related to hypoestrogenism). The majority of patients presenting with amenorrhea and anorexia have some degree of low bone density, and bone mass can decline by as much as 2% to 6% per year during periods of undernutrition. Unfortunately, BMD does not always normalize after restoration of a healthy body weight and menstrual cyclicity, often because peak bone mass was not achieved during the adolescent and young adult years. The declines in bone density have been shown to correlate with the duration of anorexia and the age of onset of the disease. Treatment of anorexia-associated osteopenia appears to require more than hormone replacement, likely because the osteopenia results from various nutritional and hormonal factors and not just estrogen deficiency. For example, patients who received recombinant human insulin-like growth factor-1 (IGF-1) in combination with oral contraceptives had higher BMD than those who received oral contraceptives alone.
Males account for only 5% of the anorexic population, but the diagnosis of AN in males is often delayed because the signs of reproductive dysfunction are harder to identify. Anorexia in men is strongly associated with depression and other underlying psychiatric disturbances, although the rate of comorbid substance abuse is higher than it is in women. Other features more prevalent in males with anorexia include a history of obesity, sexual identity concerns, and excessive exercising. Boys are at higher risk for malnutrition-induced delays in growth and pubertal development, because they enter puberty later than girls. Studies in males with anorexia have shown abnormalities in the hypothalamic–pituitary–gonadal axis similar to those in women. Serum testosterone concentrations are often below normal, as are mean 24-hour LH concentrations. GnRH-stimulated gonadotropin levels are also lower than those of normal control male subjects. Unlike gonadotropin levels in anorexic females, basal LH and FSH do not correlate with BMI (and degree of weight loss) in males with anorexia. This hypogonadotropic hypogonadism is likely the result of severe caloric restriction itself because caloric restriction in obese men had similar effects on sex steroid and gonadotropin levels.
States of Undernutrition: Bulimia Nervosa
Bulimia nervosa (BN) is another eating disorder characterized by irregular feeding patterns, specifically binge eating, in individuals of normal body weight, many of whom aspire to body weight far below normal. These episodes of binge eating are followed by periods of self-induced vomiting, laxative abuse, or extreme exercise, all driven by an abnormal body image. BN affects approximately 2% of the general female population and approximately one third of patients with BN who present for treatment have a history of anorexia. Both anorexia and bulimia are more common in patients with coexisting low self-esteem, depression, and anxiety. Disturbances in the reproductive axis can be seen in patients with bulimia, but they do not define the condition, as in anorexia. Amenorrhea occurs in 30% of patients with bulimia. Approximately half of women with bulimia show hypogonadotropic hypogonadism and no evidence of follicular development associated with decreased LH pulse frequency, whereas others have normal gonadotropin secretion and normal follicular development but impaired luteal-phase progesterone levels. Patients with purging behaviors showed more severe reductions in LH responses to GnRH infusion than nonpurging patients with bulimia and control subjects. As in women with anorexia, serum cortisol is higher in bulimic women than in control subjects, possibly related to increased CRH stimulation.
States of Overnutrition
Overnutrition and obesity pose a much greater threat to human health in most parts of the world than food scarcity. Associated morbidities, including diabetes, obstructive sleep apnea, cardiovascular disease, and osteoarthritis, often remain undiagnosed until later in adulthood, although their foundations are established in young adulthood. However, men and women of reproductive age can have obesity-associated reproductive dysfunction that brings them to medical attention much sooner than these other conditions. For example, the most common endocrinopathy in reproductive-age women is PCOS, which affects 5% to 10% of women in this age group and is associated with reproductive as well as cardiovascular and cosmetic complications. Many patients with PCOS are obese, and adiposity correlates linearly with testosterone levels in these women. Weight reduction can restore menstrual regularity and, therefore, fertility in many patients with PCOS. Both lean and obese women with PCOS show rapid LH pulse frequencies, but increasing body weight is associated with decreases in LH pulse amplitude in women with PCOS. This neuroendocrine dysfunction is a product of and contributes to hyperandrogenemia, thus creating a vicious cycle in women with PCOS.
Obesity itself in the absence of PCOS-associated hyperandrogenemia also affects the hypothalamic–pituitary axis. Historical analysis of menstrual cycle data shows a higher percentage of menstrual abnormalities or hirsutism in women who were heavier. The number of menstrual cycles with evidence of luteal activity was lower in overweight or obese women than in normal-weight participants in the Study of Women’s Health Across the Nation (SWAN) study. Women with BMI of 25 kg/m 2 or greater also had statistically longer follicular phases and shorter luteal phases. Consistent with these population studies, detailed hormonal sampling studies have shown that excess body weight suppresses gonadotropins. LH amplitude was reduced by 50% in morbidly obese women (mean BMI, 48.6 kg/m 2 ) with regular cycles studied before gastric bypass surgery, although pulse frequencies did not differ. Nevertheless, the marked attenuation of pulse amplitude resulted in a lower mean LH compared with normal-weight control subjects. This reduction in LH drive also results in deficient luteal stimulation and corpus luteum progesterone production, as reflected by lower urinary excretion of the progesterone metabolite pregnanediol glucuronide, illustrated in Fig. 20.13 . Plasma E 2 and androstenedione concentrations are lower in obese women with regular cycles, although E 1 does not change with body weight in most studies.
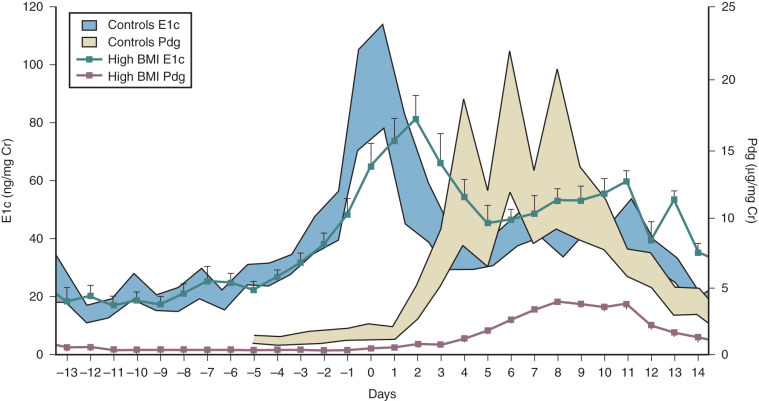
Multiple factors are likely responsible for these obesity-associated neuroendocrine changes. The response to exogenous physiological-dosed GnRH was inversely related to body weight in women with PCOS but not in normally cycling women, suggesting that obesity may blunt the pituitary response to GnRH. Insulin may also interfere with pituitary gonadotropin secretion, although no clear relationship between LH and insulin has been identified. However, hyperinsulinemia clearly suppresses hepatic secretion, thereby increasing free E 2 levels. Free E 2 may exert more negative feedback on the pituitary because it is more biologically active, thus decreasing gonadotropin production. Leptin, an adipose-derived hormone, increases hypothalamic GnRH pulsatility and pituitary gonadotropin release, although obesity is often associated with leptin resistance. Leptin also inhibits granulosa cell responses to hCG and may interfere with oocyte maturation. Thus the exact relationship of leptin to the neuroendocrine changes described earlier is not fully understood. Similarly, the role of other adipose-derived substances, including TNF-α and interleukin-1β, is an area of active investigation because both have the ability to reduce pituitary response to GnRH as well as to impair corpus luteum function.
Obesity in men is also associated with reductions in circulating gonadotropin and sex steroid concentrations. The hyperinsulinemia-associated decrease in hepatic SHBG decreases binding affinity for testosterone and results in greater aromatization of testosterone to E 2 . The subsequent increase in free E 2 and the reduction in the testosterone-to-E 2 ratio have important implications for male sexual function. Total testosterone is lower in obese men compared with normal-weight men, but free testosterone does not change. Mean LH, LH pulse frequency, and LH pulse amplitude are suppressed in severely obese men (BMI > 35), again reflecting the negative feedback effects of elevated free E 2 . More moderate degrees of adiposity (BMI 30 to 35) do not affect gonadotropin secretion. These alterations in the hypothalamic-pituitary-gonadal axis can decrease sperm concentrations. Importantly, reducing hyperinsulinemia through weight loss allows for restoration of normal testosterone and LH levels.
The restoration of the gonadal axis is independent from the variation of the macronutrition components in the weight loss diet.
Effects of Exercise on the Neuroendocrine Components of the Reproductive Axis
Excessive exercise in the absence of eating disorders can result in hypogonadotropic hypogonadism due to hypothalamic dysfunction (“athletic amenorrhea”). The clinical manifestations of the reduced GnRH activity are varied and may include amenorrhea, oligomenorrhea, LPD, or anovulation. Menstrual cycle length is not a good marker of ovarian function in athletic women because it may be normal during cycles characterized by anovulation or LPD. In addition, one woman’s cycle will differ over time, depending on the volume of her exercise and other environmental factors. Exercise-associated menstrual dysfunction has been reported in women engaged in a variety of athletic activities, including long-distance running, rowing, skiing, tennis, gymnastics, ballet, fencing, and volleyball. Dancers and long-distance runners have especially high rates of menstrual dysfunction (60% to 70%), but divers, cheerleaders, and gymnasts also have rates of amenorrhea (22%) that far exceed those in the general population. Even recreational athletes are susceptible to exercise-associated menstrual dysfunction; 78% of such women had LPD or anovulation in at least one of three menstrual cycles.
Risk factors for exercise-associated menstrual irregularities include the volume of training, eating behaviors, and gynecologic age. Studies of distance runners have illustrated the effect of exercise volume on menstrual function, with the prevalence of amenorrhea increasing from 3% to 60% as weekly training mileage increased. Surveys of eating behaviors among female collegiate athletes show relatively low rates of diagnosed eating disorders among this population (3.3% with AN; 2.3% with BN). Importantly, however, almost one third may be “at risk” for eating disorders, based on assessments of their attitudes toward eating, and these athletes are the ones most likely to report menstrual irregularities or bone injuries. Although all menstruating females are susceptible to the disruptive effects of exercise on the reproductive axis, those who are younger and closer to the time of menarche (i.e., lower gynecologic age) are more sensitive, regardless of body size, body composition, training mileage, or years of training. This sensitivity of the hypothalamic–pituitary–gonadal axis appears to be maximal in the first 15 years after the onset of menses and wanes thereafter.
Exercise-induced menstrual dysfunction is characterized by low gonadotropin and E 2 concentrations. Whereas athletic women with normal cycles had 20% to 30% reductions in LH pulse frequency compared with regularly cycling sedentary women, amenorrheic athletes experienced an additional 20% reduction in LH pulse frequency, as shown in Fig. 20.14 .
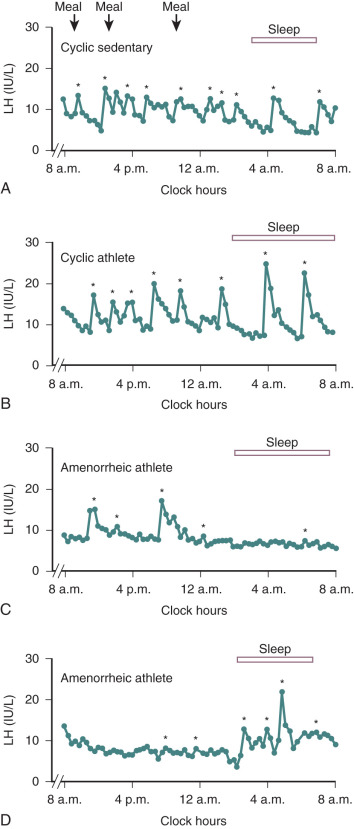
Reductions in LH pulse amplitude and loss of sleep-associated changes in LH pulsatility also occur in the amenorrheic group. FSH secretion is impaired during the follicular-luteal phase transition. These gonadotropin changes may be subtle enough that menses remain regular, but they likely contribute to the high rates of anovulation (12%) seen in recreational athletes. Ultimately, ovarian steroid production is reduced in the absence of appropriate gonadotropin stimulation, and corpus luteum function is impaired. The former is reflected in Fig. 20.15 (top) by a reduction in urinary E 1 (E 1 G) excretion in amenorrheic athletes, whereas Fig. 20.15 (bottom) shows urinary excretion of the progesterone metabolite pregnanediol glucuronide (PdG) in these women. Reductions in PdG define LPD and characterize 43% of the menstrual cycles in regularly cycling athletic women and can contribute to infertility.
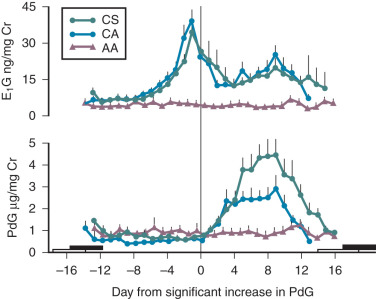
Provision of exogenous GnRH to women with exercise-induced amenorrhea illustrated that the etiology of this disorder is reduced hypothalamic GnRH secretion, rather than impaired pituitary or ovarian responsiveness. Submaximal graded doses of GnRH (2.5 µg, 5 µg, 10 µg, 25 µg) elicited exaggerated LH responses in athletic women compared with sedentary control women, whereas supraphysiologic doses of GnRH (100 µg) in amenorrheic athletes resulted in greater LH secretion compared with regularly menstruating athletic women.
Exercise also causes hypogonadotropic hypogonadism in men, with reductions in both gonadotropins and total and free testosterone levels that correlate with the intensity of endurance exercise. High-mileage endurance exercise can also affect sperm count, density, motility, and morphology on semen analysis. Unlike endurance exercise, resistance exercise is associated with transient increases in androgens and dehydroepiandrosterone sulfate in men. Because hypogonadism in males is less often recognized by both medical providers and patients themselves, literature on the effects of exercise on the male reproductive axis is more limited.
Physical activity is associated with hypogonadotropic hypogonadism in some, but not all, female athletes. A prospective study of previously eumenorrheic women who engaged in distance running (65 miles/week) at or above the lactate threshold for 12 months or longer detected no significant changes in menstrual cycle length, follicular and luteal phase length, or integrated serum progesterone and E 2 concentrations compared with control women who were not exercise-trained. Body weight, percentage of body fat, lean body mass, and caloric intake did not change during the 12-month training period, and none of the women had oligomenorrhea or amenorrhea. Only luteal phase length was shorter in the group exercising above the lactate threshold. This stability of the reproductive axis occurred despite significant increases in oxygen consumption in the training groups. Importantly, the women in this study had few risk factors for exercise-induced reproductive dysfunction because their mean gynecologic age was 17 years and they were carefully screened for coexisting psychiatric disease or eating disorders. Nevertheless, critics of this study note that the trained women may have been exercising enough at baseline to alter reproductive hormone secretion, although there was little evidence that intensifying their exercise adversely affected their reproductive function.
The mechanisms whereby some women lose menstrual cyclicity when they initiate an exercise regimen are complex. Loss of body fat with intense exercise, with or without an underlying eating disorder, has long been thought to explain exercise-induced amenorrhea. Maintenance of a minimal percentage of body fat and a minimal weight for height was believed to be necessary to prevent menstrual dysfunction or restore menses in athletic women experiencing amenorrhea. However, this theory has been disproved in multiple studies, none of which has been able to differentiate normally cycling athletes from oligomenorrheic or amenorrheic athletes by anthropometric features alone. In addition, the changes in LH pulsatility are evident within 5 days, which is much sooner than changes in adiposity appear.
An imbalance between caloric intake and the amount of energy expended with exercise more likely explains the reduced hypothalamic GnRH drive observed in women with exercise-associated menstrual dysfunction. Energy availability can be defined by the difference between energy intake and energy expenditure, with exercise being one component of energy expenditure. When energy availability is low, reproductive function suffers, as do thermoregulation, growth, and cellular maintenance. Detailed nutritional assessments of athletic women who were expending 900 to 1000 kcal/day exercising showed that their daily caloric intake was similar to that of sedentary women. Thus energy availability was significantly lower in the former, although none of these athletes had been formally diagnosed with an eating disorder. Fat and protein accounted for a smaller percentage of calories in the athletic women, and the athletic women with amenorrhea consumed 50% less fat than their regularly menstruating counterparts. This negative energy balance was reflected in insulin–glucose dynamics, with amenorrheic athletes showing marked hypoinsulinemia throughout the 24-hour observation period and glucose levels that were 10% lower. Women with more significant hypoinsulinemia had the lowest dietary fat intake and experienced greater reductions in LH pulse frequency.
These effects of energy availability on hypothalamic GnRH secretion occur relatively quickly. When a negative energy balance is created experimentally by restricting calories, increasing expenditure, or combining the two, declines in LH pulse frequency occur within 1 week. Detailed studies in women whose energy availabilities were gradually decreased while they maintained a constant energy expenditure of 15 kcal/kg lean body mass (LBM)/day (kcal/kg LBM × day) illustrated that 30 kcal/kg LBM × day is the threshold of energy availability required to maintain normal reproductive function. When energy availability falls below this threshold, LH pulse frequency declines and amplitude increases, as illustrated in Fig. 20.16 .
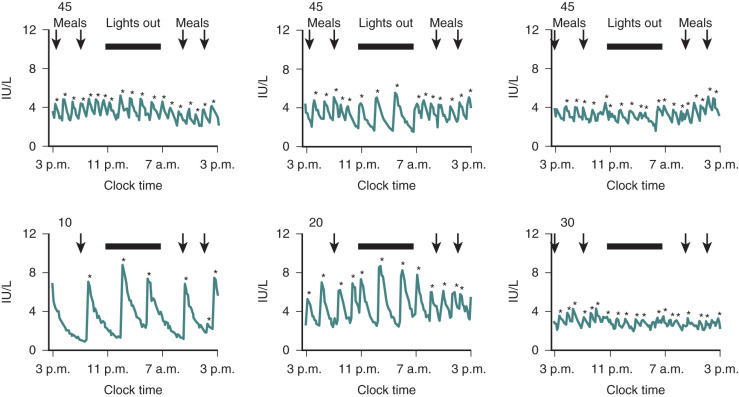
The sensitivity of GnRH neurons to energy availability appears to decline with increasing gynecologic age, suggesting that hypothalamic centers become desensitized to these metabolical signals over time. Caloric restriction combined with vigorous daily aerobic exercise altered LH pulse frequency in adolescents but not in adults; these changes appeared within 5 days of energy deprivation, as shown in Fig. 20.17 . Correction of this negative energy balance in amenorrheic exercising monkeys who continued vigorous training was achieved with overnutrition, and the rapidity of recovery was directly related to energy consumption.
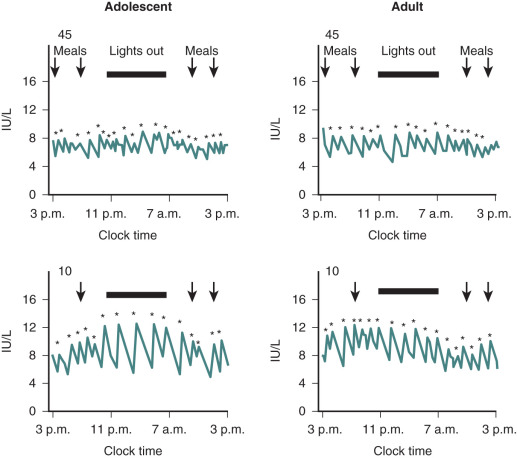
These experimental data suggest a role for a peripheral marker of adequate energy stores that can also act centrally to regulate the reproductive axis. Derived principally from adipose tissue, leptin is one such hormone whose receptors have been identified in the hypothalamus, pituitary, ovary, and testes. Its effects are stimulatory in the CNS and inhibitory at the gonads. Leptin concentrations in amenorrheic athletic women are significantly reduced compared with those in eumenorrheic athletic women, and the normal diurnal pattern of leptin secretion is lost in the former. Opposing the actions of leptin, ghrelin is an orexigenic hormone that stimulates the release of NPY and agouti-related protein from the arcuate nucleus of the hypothalamus. Exercise does not appear to alter ghrelin levels in the short term. Chronic exercise over 1 year in overweight sedentary women was associated with increases in ghrelin when the exercise was accompanied by weight loss. Not surprisingly, amenorrheic athletes had 85% higher levels of ghrelin than women who were ovulatory and sedentary, ovulating and exercising, or exercising with LPD. Thus both leptin and ghrelin are likely involved in the metabolical signaling cascade that mediates the reproductive effects of exercise.
Exercise may also impair the hypothalamic–pituitary–gonadal axis in some women by acting as a chronic stressor and activating the HPA axis. This activation mobilizes fuel stores in an attempt to maintain metabolical homeostasis. Ultimately, however, this HPA activation impairs other hypothalamic–pituitary axes, including the hypothalamic–pituitary–gonadal axis, through changes in CRH, proopiomelanocortin, ACTH, endogenous opiates, adrenal corticosteroids, or neurotransmitters. Both regularly menstruating and amenorrheic female athletes demonstrated higher plasma cortisol levels compared with regularly menstruating nonathletes with preservation of a diurnal pattern. The degree of hypercortisolism correlated negatively with the magnitude of reduction in LH pulse frequency. Administration of carbohydrates during prolonged exercise was shown to attenuate these increases in cortisol in both rodents and humans, suggesting that this HPA axis activation may occur in response to inadequate energy availability and that dietary energy restriction is more disruptive to the reproductive axis than energy expenditure through exercise.
Alterations in the somatotroph axis also occur as a result of exercise-associated metabolical changes. Both GH pulse frequency and 24-hour mean GH concentrations are augmented in amenorrheic athletes, changes that are also observed in states of nutritional deficiency, such as AN. These increases in GH levels do not effect a rise in circulating serum IGF-1, which directly stimulates LH release in rodent studies. This lack of increase in IGF-1 likely reflects the increases in its binding protein (IGF binding protein-1 [IGFBP-1]), which is the most metabolically responsive IGF-binding protein. In the prevailing hormonal milieu associated with exercise (low endogenous insulin, elevated cortisol), IGFBP-1 increases and serves as another peripheral signal of fuel shortage, slowing hypothalamic GnRH release. Unlike IGFBP-1, the concentrations of other GH-binding proteins (GHBPs) fall 35% in amenorrheic athletes and are unchanged in regularly cycling athletes, demonstrating the role that nutrient availability plays in the regulation of hepatic GHBP synthesis. Exercise-associated variations in circulating catecholamine and free fatty acid levels may also contribute to this complex regulation of hypothalamic-pituitary GH secretion.
Given its role in metabolism, it is not surprising that thyroid hormone is affected by the hypothalamic dysfunction that occurs with exercise of moderate intensity. Energy imbalance as a result of carbohydrate restriction causes triiodothyronine (T 3 ) to decline, the result of reduced 5′-diodinase activity. In men, increased energy expenditure through moderate exercise results in a rise in reverse T 3 and a fall in serum free T 3 , changes similar to those seen in the “low T 3 syndrome.” Exercise in women is associated with significant reductions in T 4 and T 3 , although only amenorrheic athletic women have alterations in free T 3 and reverse T 3 levels, both of which are significantly decreased compared with regularly menstruating athletes or sedentary women. Mean 24-hour TSH levels are not affected by this reduction in thyroid hormone negative feedback. Thyrotropin-releasing hormone (TRH) deficiency with exercise could explain these differences in women. Alternatively, the nocturnal hypermelatoninemia observed in amenorrheic athletes could modulate thyroid hormone levels because melatonin secretion from the pineal gland reduces serum thyroid hormone levels in animals and humans. Finally, some studies suggest a genetic predisposition that contributes to the susceptibility to inhibition of the reproductive axis by stressors such as exercise and nutritional deficits as well as psychologic stressors.
In summary, multiple metabolical signals are clearly involved with signaling to the GnRH pulse generator. Exercise can temporarily alter these metabolical signals if it creates an imbalance in energy availability. Such an imbalance can be prevented by supplementing dietary intake. Although most female athletes do not intentionally restrict their intake, few consume the additional calories needed to maintain a healthy energy balance. The exact nature of these interactions is complex and continues to be the subject of multiple investigations but appears to be independent of body weight.
Long-Term Complications of the Hypoestrogenism Associated With Exercise
The complications associated with hypoestrogenism and exercise include infertility, lipid abnormalities, and premature bone loss. Infertility results from poor or absent ovarian follicular development, impaired ovulation, or insufficient luteal-phase progesterone concentrations to support implantation. Amenorrheic athletes frequently have elevated low-density lipoprotein cholesterol levels and endothelial dysfunction, despite their lack of body fat and low dietary fat intake. Although the long-term effects of these cardiovascular risk factors in this population are not known, the adverse effects of amenorrhea on the skeletal system in young female athletes are well established. Estimates of the prevalence of osteopenia among female athletes range from 22% to 50%, whereas the prevalence of osteoporosis is much lower (0% to 13%). Reductions in BMD are the result of increased bone resorption secondary to estrogen deficiency and decreased bone formation resulting from reduced energy availability. Exercise itself may have differential effects on BMD, depending on the site examined. BMD at weight-bearing sites containing predominantly cortical bone, such as the femoral neck, was normal or increased in adolescent ballet dancers, whereas BMD was reduced at sites with predominantly trabecular bone, such as the spine and ribs. Sedentary amenorrheic girls with anorexia whose percentage of body fat was similar to the ballet dancers had lower BMD at all sites compared with regularly menstruating girls. Thus weight-bearing exercise may offset the adverse effects of exercise-associated hypogonadism on cortical bone. The skeletal effects of milder degrees of reproductive dysfunction, such as LPD, are unknown, and increasing evidence suggests that the declines in BMD associated with exercise-induced hypogonadotropic hypogonadism may not be reversible.
The American College of Sports Medicine uses the term “the female athlete triad” to refer to the association of menstrual dysfunction, disordered eating, and osteoporosis in female athletes. Fig. 20.18 illustrates the importance of optimal energy availability for both reproductive and skeletal health.
Caloric intake that is adjusted for the energy expended during exercise maintains normal hypothalamic–pituitary–gonadal function (eumenorrhea) while also providing nutrients for normal bone formation. Athletes who maintain such homeostasis often have BMD 5% to 15% above those of age-matched individuals. However, even mild but sustained reductions in energy availability can induce subtle degrees of menstrual dysfunction. Eventually, such dysfunction impairs estrogen-mediated bone resorption and BMD begins to fall below that expected. Not surprisingly, eumenorrheic athletes have higher BMD than amenorrheic athletes, and eumenorrheic athletes with disordered eating but no formal eating disorder have lower BMD.
Identifying adolescents and women with the female athlete triad can be difficult, especially when overt amenorrhea has not developed. Even in the absence of a clinical eating disorder, restrictive eating behaviors should raise concern and should be investigated further, potentially with the help of a mental health professional. A history of multiple stress fractures or fractures with minimal trauma might also be the first clinical manifestations of nutritional deficits. Health care providers should be especially mindful of menstrual dysfunction in adolescent females engaged in sports where leanness is emphasized (i.e., gymnastics, cheerleading, diving). Such athletes should be encouraged to optimize their nutrition and vitamin D status to prevent exercise-induced menstrual dysfunction. Exercise-associated menstrual dysfunction is a diagnosis of exclusion, however, and women should still be evaluated for pregnancy, hyperprolactinemia, thyroid dysfunction, premature ovarian failure, and anatomical abnormalities of the uterus, even if the history is consistent with exercise as the cause of oligomenorrhea or amenorrhea.
Treatment
Treatment of adolescents and adults with exercise-associated reproductive dysfunction requires a modification in diet or exercise behavior to increase energy availability, an intervention that is often difficult to implement. Such nonpharmacologic approaches are also more effective at increasing bone density than is hormone replacement therapy (HRT). In the absence of weight gain, HRT has not been consistently effective at increasing BMD in patients with exercise-induced hypogonadotropic hypogonadism, likely because it does not correct the abnormal metabolical milieu that may be contributing to premature bone loss. In addition, many athletes refuse to take estrogen because of the associated weight gain. HRT may be indicated in a woman with continued loss of BMD on serial bone densitometry scans, despite adequate nutritional intake and body weight. Bisphosphonates are not used for the bone loss associated with exercise-induced amenorrhea because of their long residence time in bone and the harm they may cause to future developing fetuses. Recombinant IGF-1 used in combination with HRT in the form of oral contraceptive pills may be better than oral contraceptives alone at increasing BMD, but this is not currently used clinically.
Infertility related to exercise-induced alterations of the hypothalamus can be treated with exogenous gonadotropins, but more conservative interventions, such as restoration of body weight, are much preferred. Women with low body weight and borderline nutritional status before conception may not augment their intake enough during pregnancy to sustain normal fetal growth, thus placing them at higher risk for spontaneous fetal loss. Progesterone can be prescribed for those women with LPD that does not correct, despite increases in energy intake or decreases in exercise.
Stress and Functional Hypothalamic Amenorrhea
Undernutrition, overnutrition, and exercise are well-recognized causes of functional hypothalamic amenorrhea (FHA), which is defined by hypothalamic hypogonadism in the absence of any structural hypothalamic or pituitary abnormality. The mediators of this hypothalamic dysfunction are multiple and likely include insulin, leptin, ghrelin, CRH, and cortisol. FHA accounts for 34% of cases of secondary amenorrhea in adults, making it the most common cause of this disorder. FHA also exists in women who do not meet the criteria for an eating disorder and do not exercise; such cases are often attributed to stress or are termed idiopathic. Life-threatening stress (i.e., experiencing refugee or concentration camps, wars, air raids) has long been known to cause amenorrhea. In the majority of women, it resolves once environmental conditions improve. Less severe stressors also contribute to menstrual dysfunction in some women. Characterization of women with such stress-associated menstrual dysfunction is complicated by the heterogeneity of the condition.
Stress is difficult to define objectively, but Selye noted in 1939 that it was accompanied by activation of the HPA axis and that it reduced reproductive capacity. This physiologic response makes sense teleologically because it allows for preservation of adrenocortical function in the case of emergency at the expense of gonadal activity. Acute stress results in a brief increase in LH and testosterone, possibly as a result of a decrease in the metabolical clearance rate rather than a change in the secretory rate. However, this initial stimulatory phase is short-lived and is not nearly as clinically relevant as the response to prolonged stress, which has consistently been shown to inhibit hypothalamic GnRH secretion, to interfere with GnRH-induced LH production, and to impair gonadal response to LH and FSH. Stress-related increases in CRH, proopiomelanocortin, ACTH, endogenous opiates, adrenal corticosteroids, or neurotransmitters may modulate these central and peripheral effects on the reproductive axis. The interactions of each of these factors with each other and with the hypothalamus, pituitary, and gonads are extremely complex, as shown in Fig. 20.19 , and they are therefore difficult to distinguish experimentally.
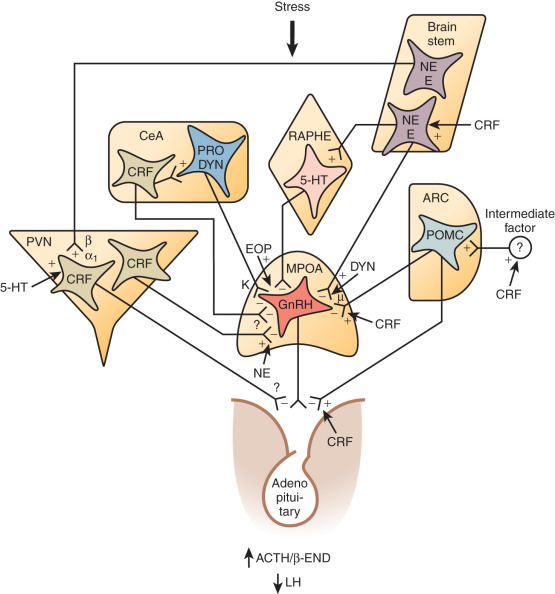
The decreased GnRH activity characteristic of FHA reflects CNS and hypothalamic responses to perceived or actual stressors. Different stressors elicit different neuromodulatory responses in different people, and the magnitude of these responses may be determined early in life by events that permanently influence the sensitivity of the HPA axis. The severity of patients’ self-reported psychosomatic disturbance has been shown to correlate with the degree of gonadotropin suppression. Most women are able to maintain normal menstrual cycles throughout stressful life events, periods of weight loss, and periods of intense exercise. In those who have amenorrhea, some degree of menstrual irregularity often precedes the complete cessation of menses, whereas women with amenorrhea related to Cushing syndrome, hypothyroidism, Sheehan syndrome, or pituitary tumors usually have no history of menstrual dysfunction. Similarly, athletes who have amenorrhea during periods of intense exercise often have a history of menstrual irregularity. Clearly, in some individuals, the hypothalamic–pituitary–gonadal axis is more sensitive to disturbances in homeostasis than it is in others. Men are also susceptible to stress-associated reproductive compromise, but identifying functional declines in GnRH pulsatility is quite difficult in males. Asthenospermia (decline in sperm motility) may be the mildest manifestation of altered GnRH drive but is usually only diagnosed in the workup of infertility.
Gonadotropin secretion in women with FHA is inappropriately low, but the pattern of secretion varies during the course of illness. Studies in women with FHA who were of normal weight for height, who were not being treated for an eating disorder, and who were not exercising excessively at the time of sampling showed low-frequency LH secretion in 43%, low-frequency and low-amplitude secretion in 27%, low-amplitude secretion in 8%, apulsatile secretion in 8%, and an unclassified pattern in 14%, as shown in Fig. 20.20 .
Related posts:
 Gonadotropin Hormones and Their Receptors
Gonadotropin Hormones and Their Receptors
 Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
 Structure, Function, and Evaluation of the Female Reproductive Tract
Structure, Function, and Evaluation of the Female Reproductive Tract
 Endocrine Disturbances Affecting Reproduction
Endocrine Disturbances Affecting Reproduction
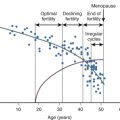 Medical Approaches to Ovarian Stimulation for Infertility
Medical Approaches to Ovarian Stimulation for Infertility
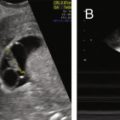 Pelvic Imaging in Reproductive Endocrinology
Pelvic Imaging in Reproductive Endocrinology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


