10.1
Introduction
The biological role of phosphorus in humans and other organisms’ physiology is diverse. Phosphorus is essential for the proper function of a multitude of systems. In addition to its critical role in skeletal mineralization, phosphorus is essential for a variety of biochemical processes . Phosphorus plays an integral part in energy homeostasis, enzyme function, and cell membrane integrity . Significant hypophosphatemia can result in skeletal, hematopoietic, muscle, or cardiac dysfunction . Phosphorus is a key substrate in bone and appropriate concentrations are required for normal mineralization to occur . Calcium and phosphorus are incorporated into the skeleton primarily in the form of hydroxyapatite. Conditions resulting in chronic hypophosphatemia are associated with abnormal mineralization and manifest as rickets in children or osteomalacia in adults . Mineralization defects can occur in spite of normal concentrations of calcium, 1α,25-dihydroxyvitamin D 3 [1α,25(OH) 2 D 3 ], and parathyroid hormone (PTH) when phosphorus concentrations are insufficient. Diseases such as X-linked hypophosphatemic rickets (XLH), autosomal dominant hypophosphatemic rickets (ADHR) , and tumor-induced osteomalacia (TIO) are disorders characterized histologically by widened osteoid seams as a result of defective mineralization due to hypophosphatemia . The majority of patients with these disorders have normal or near normal concentrations of calcium and PTH . Although additional factors may be involved, this points to the essential role phosphorus plays in normal skeletal biology.
Many factors affect the absorption/reabsorption of phosphorus in the intestine and kidney that ultimately influence concentrations in the blood ( Table 10.1 ). Classically, the major hormones involved are considered to be 1α,25(OH) 2 D 3 and PTH. However, it has become clear that phosphaturic peptides also play an important role in disorders of phosphate homeostasis and skeletal mineralization . In this chapter, we will discuss the role of 1α,25(OH) 2 D 3 and PTH as well as the role of the phosphaturic peptides fibroblast growth factor 23 (FGF23), secreted frizzled-related protein 4 (sFRP4), matrix extracellular phosphoglycoprotein (MEPE), and fibroblast growth factor 7 (FGF7) in abnormal, and normal phosphate homeostasis.
| Increase | Decrease |
|---|---|
|
|
10.2
Phosphorus homeostasis
The majority of phosphate in humans is found in the skeleton with the remainder distributed in other tissues and the extracellular space. Phosphorus balance is primarily determined by intestinal absorption and renal reabsorption regulated by vitamin D and PTH, respectively. Serum phosphorus concentrations reflect overall balance and the movement of phosphorus between serum and bone or soft tissue. The physiological range of serum phosphorus in adults is approximately 2.5–4.5 mg/dL. This value is higher in infants and children whose normal range is significantly higher compared to adults. Plasma phosphate concentrations are decreased by ingestion of a low-phosphate diet and increased by a high-phosphate intake. These changes are associated with a concomitant reciprocal change in plasma calcium. This, in turn, causes changes in PTH and vitamin D synthesis to restore phosphate balance. It is important to recognize that the renal and intestinal adaptations in phosphate absorption/reabsorption can also occur independent of PTH and vitamin D. Our understanding of phosphorus homeostasis is best understood in the context of its interactions with vitamin D and PTH.
10.2.1
Role of the kidney in phosphate homeostasis
In states of neutral phosphate balance, the amount of phosphate excreted in the urine is equal to the net amount of intestinal phosphate absorption. Virtually, all inorganic phosphate in the serum is filtered by the glomerulus . About 80%–90% of filtered phosphorus is reabsorbed in the kidney, primarily by the proximal tubule. The amount of phosphorus reabsorbed is greatest in the first half of the proximal tubule and exceeds that of sodium . There is evidence for further phosphorus reabsorption by the pars recta portion of the proximal tubule, particularly in the absence of PTH . Little or no phosphorus reabsorption occurs in the loop of Henle or the distal tubule. The reabsorption of phosphorus is sodium-dependent and is mediated by sodium-phosphate cotransporters (NaPi IIa and NaPi IIc) . NaPi II transporter activity is increased by ingestion of a low-phosphate diet and decreased by ingestion of a high-phosphate diet. The renal adaptation to changes in dietary phosphate intake occurs very rapidly and changes in phosphate reabsorption can occur independent of PTH. This intrinsic renal adaptation that is demonstrable in vivo and in vitro is mediated by unknown mechanisms.
PTH has been recognized as the principal hormonal regulator of renal phosphate reabsorption by the proximal tubule. However, it is important to recognize that there are additional factors that modulate the inhibition of phosphate reabsorption by PTH such as respiratory acidosis or alkalosis, volume status, catecholamines, growth hormone, and phosphatonins ( Table 10.1 ).
It is well known that PTH concentrations are exquisitely sensitive to changes in serum calcium concentrations . PTH secretion is also stimulated by high-phosphate ingestion and this effect is mediated indirectly by decreases in calcium as well as through direct mechanisms . Under normal conditions a phosphate load (intravenous or oral) will stimulate PTH release from parathyroid gland cells thus increasing renal excretion of phosphorus and maintaining normal serum phosphate concentrations. Likewise, a diet low in phosphorus will result in renal conservation of phosphorus at least partially due to a decrease in PTH secretion. The phosphaturic effect of PTH administration is the result of removal of NaPi IIa transporters from the apical brush border of renal proximal tubule cells both in vitro and in vivo ( Figs. 10.1 and 10.2 ). Chronic exposure to elevated concentrations of PTH in normal animals results in an increased fractional excretion of phosphorus and hypophosphatemia.

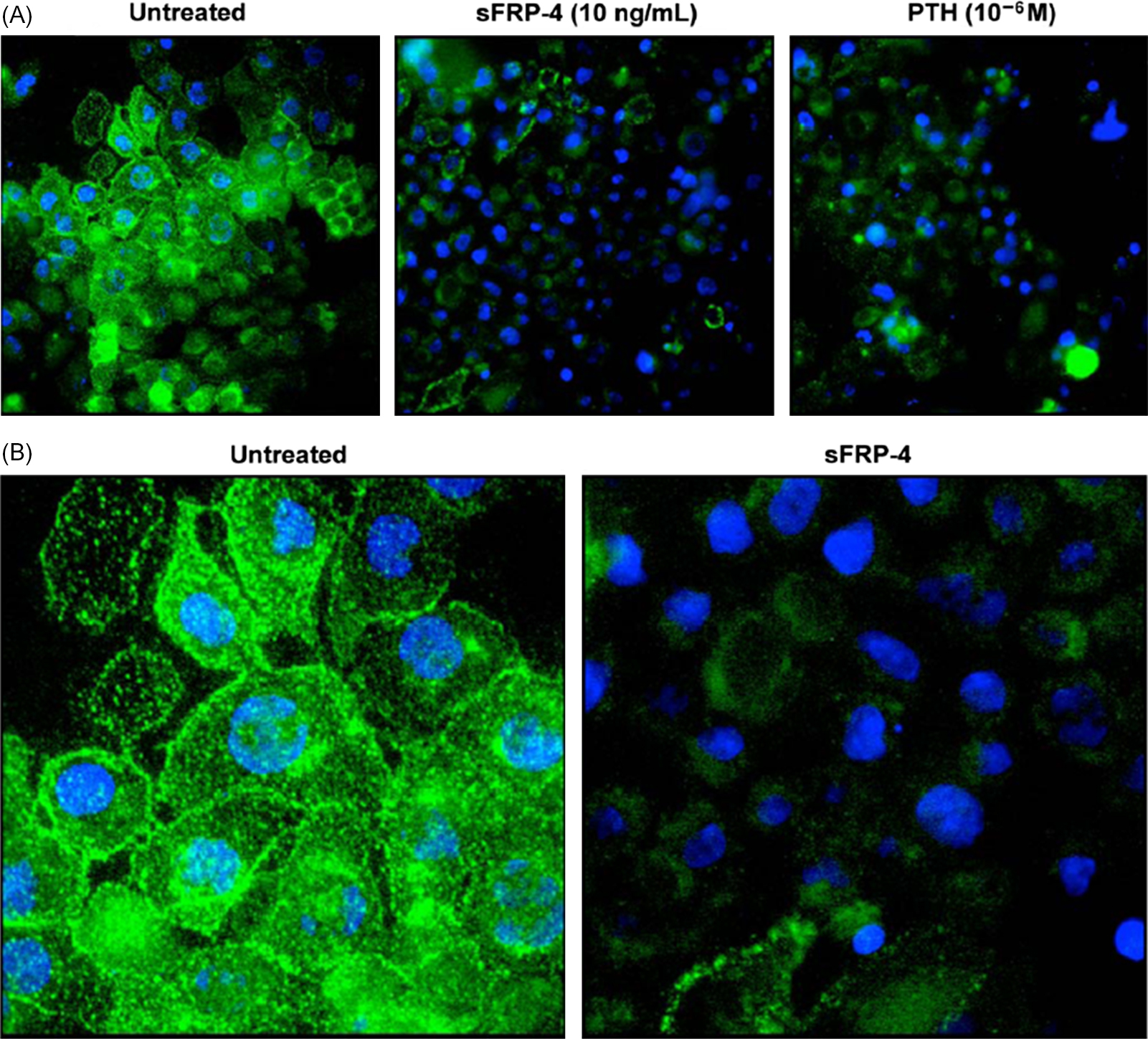
Although PTH appears to be primarily responsible for renal phosphate regulation, vitamin D also alters renal phosphate reabsorption. In vivo and in vitro studies performed by Taketani et al. demonstrated that NaPi IIa expression in renal tissues is increased by the administration of 1α,25(OH) 2 D 3 . Others have shown that the effect of 1α,25(OH) 2 D 3 on phosphate reabsorption in the kidney requires the presence of PTH .
The phosphatonins, FGF23, sFRP4, MEPE, and FGF7 also inhibit renal phosphate reabsorption in vitro and in vivo . The effects of phosphatonins on renal phosphate reabsorption will be discussed in detail later.
10.2.2
Role of the intestine in phosphate homeostasis
Phosphorus absorption in the intestine primarily takes place in the proximal small bowel with both active and passive mechanisms . The intestinal absorption of phosphorus is largely dependent on the amount of phosphorus consumed. Nonhormonal factors such as the availability of phosphorus in the gastrointestinal tract can influence serum phosphorus concentrations. Dietary calcium and other phosphate binding substances (such as sevelamer carbonate or lanthanum carbonate) will effectively reduce the amount of intestinal phosphate available for absorption.
The intestinal epithelial apical brush border contains a sodium-dependent phosphate-cotransporter, NaPi IIb. The amount of intestinal apical membrane NaPi IIb is increased in animals fed a low-phosphate diet or after the administration of 1α,25(OH) 2 D 3 . The upregulation of NaPi IIb in the intestine, while on a low-phosphate diet, is mediated by vitamin D–dependent mechanisms and is independent of PTH . Conversely, a high-phosphorus diet or elevated serum phosphorus concentrations act to decrease the expression of 25-hydroxyvitamin D-1α-hydroxylase in the renal proximal tubule cells. Decreased conversion of 25-hydroxyvitamin D 3 to 1α,25(OH) 2 D 3 will lead to a decrease in the intestinal absorption of phosphorus returning serum concentrations to the physiological range.
The important role of the NaPi IIb transporter in intestinal phosphate transport was demonstrated by tissue-specific deletion of the NaPi IIb gene in mouse intestine . NaPi II2b −/− animals had increased fecal phosphate excretion and hypophosphaturia, but serum phosphate remained unchanged. Decreased urinary phosphate excretion correlated with reduced serum levels of the phosphaturic hormone FGF23 and increased protein expression of the renal phosphate transporter NaPi IIa.
There is evidence that the intestine is capable of “sensing” luminal phosphate concentrations and influencing PTH release and the formation of unique substances that alter intestinal phosphate transport. Martin et al. demonstrated that when uremic animals fed a high-phosphate diet for 4 weeks were administered a low-phosphate diet, there was a rapid reduction in serum concentrations of PTH within 2 hours without changes in serum calcium . When uremic rats fed a high-phosphate diet were gavaged with a high-phosphate diet on the day of the experiment, PTH levels increased with only modest changes in serum phosphate during the first 30 minutes of the experiment. Phosphonoformic acid, a phosphorus uptake inhibitor, also rapidly increased PTH concentrations with no significant changes in serum phosphorus. The administration of intravenous phosphate rapidly increased PTH with no changes in serum calcium and modest increases in serum phosphorus. These data demonstrate the presence of a phosphate “sensor” in the intestine and in the parathyroid gland.
We showed rapid changes in the renal excretion of phosphate after administration of phosphate into the duodenum of intact or parathyroidectomized rats . In the intact rats the rapid increase in the fractional excretion of phosphate in the kidney occurred without changes in serum phosphate concentrations. In thyroparathyroidectomized rats, there was a similar increase in the fractional excretion of phosphate following the administration of intraduodenal phosphate. The latter experiments clearly show that increases in the renal fractional excretion of phosphate following intraduodenal phosphate infusion are independent of PTH. We showed that there was no change in serum concentrations of the known phosphaturic peptides, PTH, FGF23, and sFRP4, in both intact and parathyroidectomized rats following the infusion of intraduodenal phosphate or intraduodenal sodium chloride. Our data show that there is a sensor for phosphate in the intestine that causes an increase in the fractional excretion of phosphate in the kidney within a short period of time following the exposure of the intestinal mucosa to increased phosphate concentrations. We demonstrated the presence of a factor in the duodenal mucosa that was capable of inducing phosphaturia in the intact rats. Taken together, the data show that the intestine senses an increase in luminal phosphate concentrations and releases a substance into the circulation that inhibits renal phosphate reabsorption.
Despite our seemingly robust understanding regarding the various factors involved in normal phosphate physiology, our knowledge is incomplete. The phosphatonins have added significantly to our knowledge of phosphorus and vitamin D metabolism and bone mineralization.
10.3
Phosphatonins
Several diseases characterized by abnormal phosphorus, vitamin D, and bone metabolism have led to the discovery of factors that regulate phosphate homeostasis in physiologic and pathophysiologic conditions. Studies of inherited forms of rickets [phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX), FGF23, ectonucleotide pyrophosphatase/phosphodiesterase 1, and dentin matrix protein 1 mutations] and TIO have identified proteins that conform to the proposed definition of a phosphatonin. A phosphatonin is considered to be a circulating factor that induces phosphaturia through PTH-independent mechanisms leading to hypophosphatemia. The peptides that fulfill this definition include FGF23, MEPE, sFRP4, and FGF7. Several of these peptides also inhibit the formation of 1α,25(OH) 2 D 3 by decreasing the expression of 25-hydroxyvitamin D-1α-hydroxylase.
Prior to the identification of these phosphaturic peptides, it had long been recognized that a circulating factor was likely responsible for the hyperphosphaturia, hypophosphatemia, and rickets/osteomalacia associated with TIO and XLH. Adults with TIO present with classic symptoms of osteomalacia, including pain, weakness, and fractures or pseudofractures. Children with TIO have been described with rickets. This form of hypophosphatemic rickets/osteomalacia can be differentiated from the inherited forms of rickets in that it is acquired and can be cured if the offending tumor is removed. The observation that the hypophosphatemia and bone disease completely resolved with removal of the tumor suggested that a circulating factor, presumably arising from the tumor, caused the phosphate abnormalities. Cai et al. performed studies in which cells derived from a tumor from a patient with TIO expressed a factor that inhibited phosphate transport in opossum kidney (OK) cells . This factor was present in the supernatant of cultured tumor cells, specifically inhibited sodium-dependent phosphate transport and did not affect amino acid or glucose transport. Furthermore, when these cells were implanted into nude mice, hypophosphatemia and osteomalacia occurred.
Additional evidence that a circulating factor other than PTH could induce phosphaturia has come from studies of the mouse model of XLH. The Hyp mouse has a 3′ deletion of the gene encoding the PHEX. These mice display a phenotype consisting of hyperphosphaturia, hypophosphatemia, and osteomalacia. Studies of Hyp mice parabiosed with normal mice showed that a phosphaturia could be induced in the wild-type mouse suggesting that a circulating phosphaturic factor was present in the blood of Hyp mice . Further evidence for a circulating phosphaturic factor was offered by Nesbitt et al. who performed renal cross-transplantation studies between normal and Hyp mice . In these experiments, normal mice receiving a kidney from a Hyp mouse had normal phosphate excretion. In contrast a Hyp mouse receiving a kidney from a normal mouse continued to show phosphaturia. These studies were consistent with the concept of the existence of a humoral factor being responsible for the phosphaturia and not an intrinsic renal defect. It has been shown that alterations in PHEX in Hyp mice are responsible for altered FGF23 metabolism leading to excess circulating concentrations.
10.4
Fibroblast growth factor 23
10.4.1
Hypophosphatemic disorders with defective mineralization
10.4.1.1
Autosomal dominant hypophosphatemic rickets
FGF23 is a 251 amino acid peptide encoded on the short arm of chromosome 12 in humans. FGF23 was initially felt to play a role in the function of the ventrolateral thalamic nucleus of the brain based on in situ hybridization studies performed in mice . Shortly after this initial report of a novel fibroblast growth factor, the ADHR consortium identified missense mutations in the gene encoding FGF23 in patients with ADHR . It was speculated that the missense mutations lead to a gain of function in FGF23 and that FGF23 may be a circulating factor capable of inducing hypophosphatemia. Substitution of the arginine residues at amino acid positions 176 or 179 result in the ADHR phenotype (hypophosphatemia, hyperphosphaturia, rickets/osteomalacia, short stature, and dental abscesses) . Imel et al. reported that serum FGF23 concentrations correlated positively with disease severity in patients with ADHR, which can vary over time.
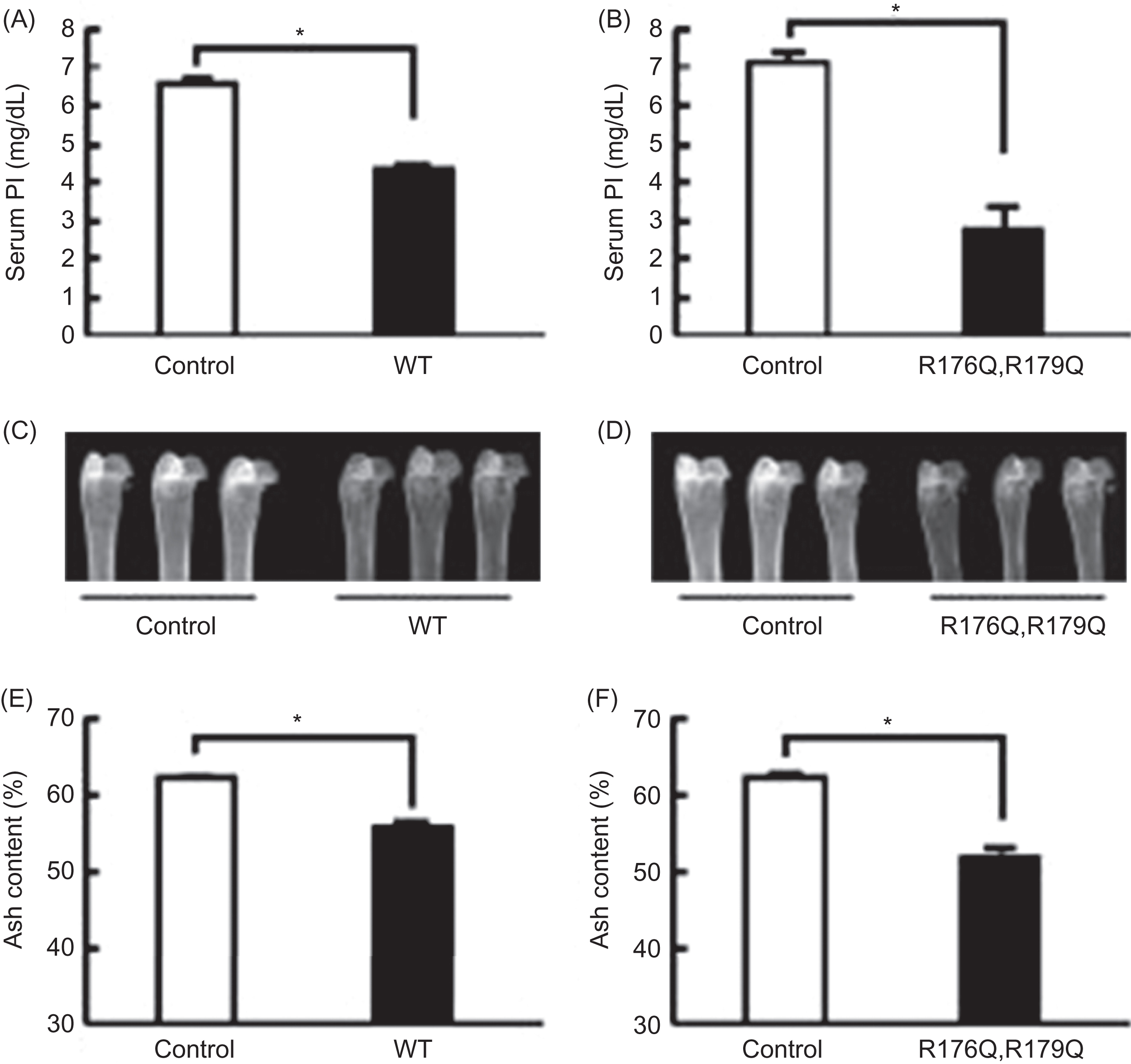
Shimada et al. demonstrated that this mutant FGF23 was resistant to proteolytic cleavage between residues 176 and 180 and was phosphaturic when administered intraperitoneally to mice . Furthermore, when cells expressing mutant or wild-type FGF23 were implanted into athymic nude mice, the animals became hypophosphatemic and had impaired bone mineralization ( Fig. 10.3 ) . Bai et al. generated transgenic mice overexpressing FGF23 to assess the role of mutant FGF23 in phosphate homeostasis . One- to two-month-old FGF23 transgenic mice exhibited hypophosphatemia, increased renal phosphate excretion, elevated alkaline phosphatase concentrations, and inappropriately low serum 1α,25(OH) 2 D 3 concentrations relative to the degree of hypophosphatemia. Femoral shortening and mineralization defects were also seen in the FGF23 transgenic mice compared to wild-type mice. These biochemical and histological characteristics are reminiscent of the findings in patients with ADHR. In addition to hypophosphatemia and inappropriately low 1α,25(OH) 2 D 3 concentrations, Saito et al. also demonstrated that mutant FGF23 reduced sodium-dependent phosphate transport in renal as well as intestinal brush border membrane vesicles . The changes in intestinal phosphate absorption were vitamin D–dependent. Several disorders associated with abnormal serum FGF23 and phosphate levels are outlined in Table 10.2 .
| Disorder | Pi concentration | FGF23 concentration | References | |
|---|---|---|---|---|
| Intact | Carboxy-terminal | |||
| XLH | Decreased | Increased | Increased | |
| ADHR | Decreased | Increased | Increased | |
| TIO | Decreased | Increased | Increased | |
| DMP1 | Decreased | Increased | Increased | |
| ENPP1 | Decreased | Increased | ||
| FAM20C | Decreased | Increased | ||
| HHM | Decreased | Increased | ||
| HLNSS | Decreased | Increased | ||
| Fibrous dysplasia | Decreased | Increased | ||
| Tumoral calcinosis | Increased | Decreased | Increased | |
| Renal failure | Increased | Increased | Increased | |
| Graves’ disease a | Variable | Variable | ||
| Ovarian cancer b | Normal | Increased | Increased | |
a FGF23 concentrations declined with antithyroid therapy.
10.4.1.2
Tumor-induced osteomalacia
TIO is an acquired disorder with many clinical and biochemical similarities to patients with ADHR and XLH. Patients exhibit signs and symptoms of rickets/osteomalacia, including bone pain, fractures, and weakness. A variety of tumor types have been described and are best characterized as phosphaturic mesenchymal tumors that have been found in virtually all regions of the body . Distinguishing TIO from inherited forms of hypophosphatemic rickets/osteomalacia can be difficult if it develops at a young age, since these tumors are notoriously difficult to locate. As previously mentioned, studies by Cai et al. demonstrated that a factor or factors secreted from cells taken from a tumor in a patient with TIO was able to inhibit sodium-dependent phosphate transport in renal tubular cells. This factor was distinct from PTH and did not alter glucose or amino acid transport in OK cells . The observation that extracts from tumors taken from patients with TIO inhibit phosphate transport in OK cells has been replicated by others .
Studies using serial analysis of gene expression demonstrated that in addition to FGF23, other phosphaturic factors, including MEPE, sFRP4, and FGF7, are also highly expressed in tumors taken from patients with TIO . The effects of FGF23 on phosphate and vitamin D metabolism have been the most characterized and the development of an assay for FGF23 in serum has allowed the assessment of the role of FGF23 in disease conditions associated with phosphate wasting. This may explain why not all patients with TIO have elevated serum concentrations of FGF23 and implies that other phosphaturic proteins may also be important in the clinical expression of this disease in some patients .
Serum concentrations of FGF23 have been measured in patients with known or presumed TIO and most but not all patients have elevated FGF23 concentrations . Furthermore, serum FGF23 concentrations decline into the normal range shortly after removal of the offending tumor . Serum phosphate and 1α,25(OH) 2 D 3 concentrations normalize within hours to days after removal of the offending tumor . However, the histological changes in bone require significantly more time to normalize .
Selective venous sampling for determination of FGF23 concentrations has been employed to confirm the location of a TIO tumor prior to surgical excision . However, this technique appears to be the most useful for confirmation of the offending tumor that has previously been identified by another imaging modality . RT-PCR, in situ hybridization, and immunohistochemical techniques have demonstrated FGF23 mRNA and protein expression in TIO tumors . These observations demonstrate that the tumor is the source of elevated circulating concentrations of FGF23.
These data provide compelling evidence that FGF23 is a causative factor inducing the biochemical and histological changes seen in TIO.
10.4.1.3
X-linked hypophosphatemic rickets
XLH is caused by mutations in the gene encoding PHEX, an endopeptidase on the X chromosome, and is characterized by hypophosphatemia, increased renal fractional excretion of phosphorus, and rickets . As previously mentioned, the Hyp mouse is the animal homologue of XLH. Studies of these mice have shown that renal phosphate wasting and the bone phenotype is due to a circulating factor and not an intrinsic renal defect . Many patients with XLH have elevated blood concentrations of FGF23 compared to normal controls . Aono et al. were able to partially rescue the phenotype of Hyp mice when treated with FGF23 neutralizing antibodies . In these experiments, repeated treatment with anti-FGF23 antibodies increased phosphorus and 1α,25(OH) 2 D 3 concentration in the serum of Hyp mice lending further evidence that FGF23 plays a central role in the hypophosphatemia and rickets observed in patients with XLH. Several investigators have suggested that FGF23 is a substrate for PHEX . Others have suggested that inactivating mutation in PHEX results in increased FGF23 production rather than lack of degradation . Either option provides an explanation for the elevated FGF23 concentrations observed in XLH as well as a mechanism for renal phosphate loss.
10.4.1.3.1
Antifibroblast growth factor 23 antibody therapy
Traditionally, hypophosphatemic disorders caused by excess FGF23 have been treated with phosphate salts and active vitamin D such as calcitriol. Although not curative, phosphate and calcitriol can improve pain, weakness, and rickets or osteomalacia caused by the hypophosphatemia. However, not all patients experienced resolution of symptoms and serum phosphorus concentrations frequently remained below the reference range for age. In addition, phosphorus and calcitriol have been shown to further increase FGF23 concentrations in healthy adults and in those with XLH that may diminish the therapeutic effect .
Aono et al. utilized the Hyp mouse model of XLH to demonstrate that anti-FGF23 antibody injections could normalize phosphorus concentrations, significantly increase 1α,25(OH) 2 D 3 and ameliorate the bone phenotype . Similar findings of prolonged normalization of serum phosphorus were seen in adults with XLH treated with recombinant human anti-FGF23 IgG monoclonal antibody . More recently, anti-FGF23 antibody (burosumab) given subcutaneously every 2–4 weeks to patients with XLH has been shown to not only correct the hypophosphatemia and inappropriate 1α,25(OH) 2 D 3 concentrations but also improve or resolve radiographic abnormalities of fractures and/or rickets in both adults and children with XLH . These data have led to the recent approval of anti-FGF23 antibody therapy by the FDA for use in children and adults with XLH. Jan De Beur et al. studied patients with TIO in whom the offending tumor could not be localized or resection was not possible . Similar to the data in patients with XLH, anti-FGF23 antibody therapy in these patients with TIO leads to increased serum phosphate and 1α,25(OH) 2 D 3 concentrations lending further evidence that anti-FGF23 therapy is effective in correcting the biochemical and clinical abnormalities in disorders of FGF23 excess.
10.4.1.4
Fibrous dysplasia/McCune Albright
Fibrous dysplasia is caused by postzygotic activating mutations in the GNAS1 gene. Fibrous dysplasia of one or more bones may be an isolated finding or associated with McCune Albright syndrome (OMIM #174800) with characteristic café-au-lait macules, peripheral precocious puberty and other related endocrine abnormalities. Some patients with these disorders also display renal phosphate wasting . The degree of phosphate wasting has been correlated with the extent of bone involvement . Phosphate wasting was not related to elevated cyclic adenosine monophosphate (cAMP) suggesting that an activating GNAS1 mutation in the kidney was not responsible for the phosphaturia. Others have reported that FGF23 is expressed in the abnormal bone of many patients with isolated fibrous dysplasia . In that study the intensity of FGF23 staining in bone tissue negatively correlated with serum phosphorus concentrations. Serum FGF23 concentrations are higher in patients with McCune Albright syndrome or fibrous dysplasia compared to age-matched controls . Furthermore, patients with renal phosphate wasting associated with fibrous dysplasia or McCune Albright have higher serum FGF23 concentrations than those without.
10.4.2
Hyperphosphatemic disorders
10.4.2.1
Tumoral calcinosis
Tumoral calcinosis is a rare condition caused in some cases by mutations in the GALNT3 gene, Klotho gene, or the FGF23 gene . The phenotype is similar despite the different genetic etiology. Biochemical findings include hyperphosphatemia, increased renal reabsorption of phosphorus, and normal or elevated 1α,25(OH) 2 D 3 . These findings are opposite to those found in patients with disorders associated with increased FGF23 activity such as XLH, ADHR, and TIO. It is interesting to note that when FGF23 concentrations are measured by a technique that identifies carboxy-terminal fragments, the concentrations are elevated. An explanation for this finding is offered by Benet-Pages et al. who demonstrated altered processing of the mutant form of FGF23 (S71G) . Expression of mutant FGF23 in HEK 293 cells resulted in the secretion of carboxy-terminal fragments of FGF23 but not intact FGF23. The intact protein was retained within the Golgi complex. Araya et al. have reported similar in vitro data . In this report, expression of mutant FGF23 (S129F) resulted in reduced detection of intact and N-terminal FGF23 by western blotting. Serum FGF23 concentrations are also elevated in their patients with tumoral calcinosis when measured with an assay that detects carboxy-terminal fragments. However, when measured using an assay that only detects intact FGF23, the concentrations were low. This suggests that biological activity of FGF23 requires an intact molecule that is not secreted in patients with tumoral calcinosis due to FGF23 or GALNT3 gene mutations. Interestingly, Ichikawa et al. reported a 13-year-old girl with severe tumoral calcinosis due to a mutation in the Klotho gene . Both intact and carboxy-terminal FGF23 concentrations were markedly elevated pointing to the importance of the coreceptor Klotho in FGF23 signaling.
Instead of mineralization defects resulting in rickets or osteomalacia, patients with tumoral calcinosis develop dramatic extraskeletal mineral deposits. A similar clinical and biochemical phenotype is apparent in FGF23 or Klotho null mice confirming that FGF23 and Klotho mutations in patients with tumoral calcinosis represent a loss of function .
10.4.2.2
Chronic kidney disease
Patients with chronic kidney disease have abnormal phosphate and vitamin D metabolism. As renal function declines, serum phosphorus concentrations increase and 1α,25(OH) 2 D 3 concentrations decrease. PTH levels are frequently elevated but insufficient to correct the hyperphosphatemia and impaired vitamin D production. Several investigators have documented increased serum concentrations of FGF23 in patients with chronic kidney disease. Initial studies were performed using an ELISA utilizing a capture and detection antibody recognizing epitopes within the carboxy-terminal portion of the protein . It was unclear whether the elevation in FGF23 was the result of increased production, decreased clearance, or the accumulation of inactive FGF23 fragments. Subsequent reports have clearly documented that intact FGF23 concentrations in serum are also elevated in patients with renal insufficiency .
It has been suggested that increased FGF23 concentrations in renal disease may represent a compensatory mechanism for hyperphosphatemia. Serum FGF23 concentrations correlate positively with serum phosphorus and with the fractional excretion of phosphorus in some patients with CKD . Also, as FGF23 concentrations increase, 1α,25(OH) 2 D 3 concentrations decline. This is not surprising since it has been shown that FGF23 acts in the renal proximal tubule to diminish 25-hydroxyvitamin D 3 1α-hydroxylase expression . A potential feedback loop may exist between FGF23 and vitamin D, since 1α,25(OH) 2 D 3 therapy in patients with CKD decreased serum FGF23 concentrations.
Decreased 1α,25(OH) 2 D 3 may lead to increased PTH production and contribute to secondary hyperparathyroidism in these patients. Kazama et al. reported that serum FGF23 concentrations were highly predictive of the development of advanced secondary hyperparathyroidism in patients receiving chronic dialysis . These investigators also found serum FGF23 levels to be predictive of their response to calcitriol therapy . The patients treated with calcitriol had significantly higher serum phosphorus and FGF23 concentration after 24 weeks of therapy. It is not clear whether the calcitriol therapy or increased serum phosphorus was directly responsible for increased serum FGF23. Another study performed in patients receiving maintenance hemodialysis found that serum phosphorus was positively associated with serum FGF23 concentrations. In this study, subjects were treated with sevelamer hydrochloride and calcium or calcium alone. The subjects receiving combined treatments had a significant reduction in serum phosphorus and FGF23, whereas subjects treated with calcium alone had no changes in either analyte . Similar findings were observed with lanthanum carbonate versus calcium in patients with stages 4–5 CKD nondialysis patients .
Elevated FGF23 has been associated with cardiac hypertrophy and increased mortality in patients with CKD . Cardiac hypertrophy appears to be mediated by FGF23 through the fibroblast growth factor receptor 4 (FGFR4) in a Klotho independent manner . Grabner et al. demonstrated in a rat model of CKD that the cardiac hypertrophy induced by elevated FGF23 can be prevented by blockade of FGFR4 and that a knock-in model with an activating FGFR4 mutation leads to cardiac hypertrophy . Although phosphate binders can reduce FGF23 concentrations in patients with CKD as noted previously, it is yet to be determined if this strategy will lead to reduced risk of cardiac events or mortality in humans.
10.4.3
Effects of fibroblast growth factor 23 in the kidney and intestine
Phosphate homeostasis is affected directly by FGF23 as a result of its inhibition of NaPi IIa and NaPi IIc cotransporter activity and indirectly by inhibition of 25-hydroxyvitamin D 3 1α-hydroxylase expression . Experiments utilizing OK cells (a proximal tubule epithelia) have demonstrated that phosphorus uptake is inhibited by FGF23 . As previously mentioned, phosphate transport in the kidney is primarily regulated by the activity of NaPi IIa cotransporters in the apical membrane. FGF23 causes internalization of NaPi IIa cotransporters and degradation in the lysosome resulting in decreased phosphate transport.
Hypophosphatemia and impaired conversion of 25-hydroxyvitamin D 3 to 1α,25(OH) 2 D 3 offer explanations for the impaired mineralization seen in the previously described disorders associated with elevated serum FGF23 concentrations.
1α,25(OH) 2 D 3 plays an important role in phosphate regulation primarily in the intestine. 25-hydroxyvitamin D 3 1α-hydroxylase converts the inactive form of vitamin D to its active metabolite 1α,25(OH) 2 D 3 , which increases phosphorus transport in the small bowel via upregulation of NaPi IIb cotransporters . XLH and TIO are both examples of hypophosphatemic disorders characterized by inappropriately low or normal 1α,25(OH) 2 D 3 concentrations relative to the degree of hypophosphatemia. This is in contrast to the marked elevation in serum 1α,25(OH) 2 D 3 concentrations that is associated with hypophosphatemia induced by dietary phosphate restriction. In fact, serum 1α,25(OH) 2 D 3 concentrations and renal 25-hydroxyvitamin D 3 1α-hydroxylase expression are decreased in animals exposed to FGF23 . In addition to correcting hypophosphatemia, 1α,25(OH) 2 D 3 also normalizes in patients with XLH treated with FGF23 neutralizing antibodies (burosumab) demonstrating the potent effect of FGF23 on 25-hydroxyvitamin D 3 1α-hydroxylase activity in humans .
Miyamoto et al. performed a set of experiments in wild-type and vitamin D receptor (VDR) null mice . The investigators injected mutant FGF23 (R179Q) that lowered serum phosphorus and 1α,25(OH) 2 D 3 concentrations. Intestinal brush border membrane vesicles of the wild-type mice showed decreased sodium-dependent phosphate transport and reduced amounts of NaPi IIb protein. In contrast, intestinal sodium-dependent phosphate transport was not affected by FGF23 (R179Q) in the VDR null mice. These data suggest that FGF23 indirectly decreases phosphate transport in the intestine by reducing serum 1α,25(OH) 2 D 3 concentrations.
10.4.4
Fibroblast growth factor 23 and Klotho
Klotho proteins are cofactors for a variety of FGF molecules, including FGF23 . Kurosu et al. demonstrated that Klotho increased the binding affinity of FGF23 with the FGF receptor . Moreover, the effect of FGF23 is diminished by the injection of wild-type mice with anti-Klotho monoclonal antibodies . Klotho mutant mice display hyperphosphatemia and elevated 1α,25(OH) 2 D 3 concentrations despite marked elevation of FGF23 . As previously mentioned, Ichikawa et al. reported a case of a 13-year-old girl with a phenotype of tumoral calcinosis resulting from a homozygous missense mutation in Klotho . Despite elevated concentrations of both intact and carboxy-terminal FGF23, she was hyperphosphatemic and developed soft tissue calcification. Taken together, these findings point to the important role of klotho as a cofactor in FGF23 action on vitamin D and mineral metabolism.
10.4.5
Effects of fibroblast growth factor 23 in bone
FGF23 has been shown to be expressed in a number of tissues, including bone. Perwad et al. measured FGF23 mRNA in the calvaria of mice fed a diet containing 0.02% or 1% phosphate. FGF23 mRNA abundance was reduced by 85% in mice fed the low-phosphate diet . In addition, FGF23 mRNA abundance was 30-fold higher in Hyp mouse calvaria, a condition known to be associated with elevated serum FGF23 concentrations. These data suggest that expression of FGF23 in bone is responsible for the changes in serum levels in patients or animals with XLH or after dietary phosphate manipulation.
It is clear that in humans and mice with altered serum FGF23 levels display distinct bone phenotypes. Hypophosphatemic disorders such as XLH, ADHR, and TIO are characterized by rickets or osteomalacia. Bone histomorphometry reveals a mineralization defect with widened osteoid seams. However, it is not clear whether these changes are due to altered phosphorus and vitamin D metabolism or if there is a direct effect of FGF23 on bone.
Several investigators have determined that FGF23 binds to various FGFRs . Yu et al. demonstrated that FGF23 binds to and activates the c-splice isoforms of FGFR1–3 and FGFR4 . Others have also shown that the binding of FGF23 to various FGFRs does so with higher affinity in the presence of the protein klotho . However, in the heart, binding of FGF23 to FGFR4 appears to be klotho-independent. FGFRs are known to play an important role in limb development, including those that appear to interact with FGF23 . Mutations in FGFR3 result in achondroplasia, hypochondroplasia, or thanatophoric dysplasia, which are characterized by various degrees of limb deformity, including shortening and bowing. Limb shortening has also been reported in FGF23 null mice . The authors also described narrowed growth plates with decreased numbers of hypertrophic chondrocytes. The ribs and vertebra of the FGF23 null mice demonstrate a marked increase in woven bone and osteoid. FGF23 null mice have similar biochemical and clinical characteristics to patients with tumoral calcinosis due to mutations in the gene encoding FGF23, including hyperphosphatemia, elevated 1α,25(OH) 2 D 3 concentrations, and extraskeletal mineralization . Chevetz et al. described a child with tumoral calcinosis due to a homozygous mutation in FGF23 (M96T). Radiographic investigation showed obvious bony abnormalities with areas of sclerosis, bowing of the distal radius, shortening of the ulna, and modeling defects in the distal femur and proximal tibia.
Although these observations suggest that FGF23 plays an important role in skeletogenesis, it is difficult to interpret in vivo evidence for direct skeletal effects of FGF23 because of its concomitant effects on phosphorus and vitamin D metabolism. However, a set of in vitro experiments performed by Shalhoub et al. using osteoblast cell culture found the combination of FGF23 and Klotho exposure resulted in impaired mineralization . This effect was mediated through FGFR1. These data suggest that there may be a direct effect of FGF23 in combination with its coreceptor Klotho on bone.
10.4.6
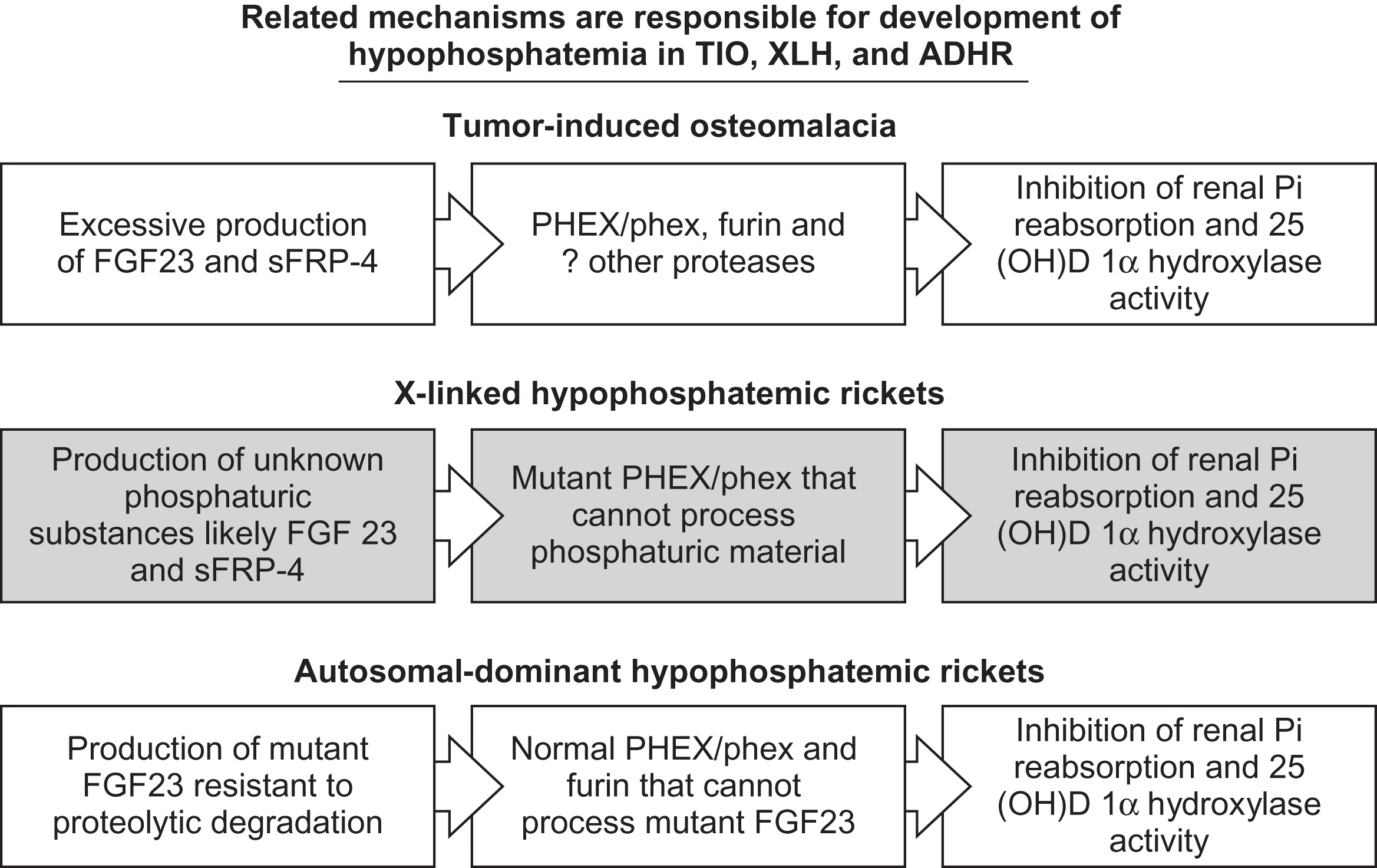
Fibroblast growth factor 23 in normal phosphate homeostasis
Significant evidence exists supporting the role of changes in serum FGF23 levels on phosphorus and vitamin D metabolism in disease states by mechanisms outlined in Fig. 10.4 . Several investigators have measured serum FGF23 concentrations after dietary manipulation of phosphorus, calcium, and/or vitamin D. Conflicting results have been reported in humans. Larsson et al. studied six healthy males for 6 days. A normal diet for 1 day was followed by 2 days of low-phosphate intake and subsequently a high-phosphate diet . However, no changes in serum FGF23 levels were noted. A larger study by Ferrari et al. evaluated 29 healthy males given a low-phosphate diet for 5 days followed by a high-phosphate diet for 5 days separated by 2 days of a normal diet. These investigators found significantly lower serum FGF23 concentrations during phosphate restriction compared to supplementation . Experiments in wild-type and VDR null mice have given additional insight into the role of FGF23 during changes in dietary phosphate intake. Wild-type mice fed a low-phosphate diet have significantly lower serum FGF23 concentrations . In the same set of experiments, it was noted that VDR null mice have very low basal FGF23 concentrations. However, when fed a rescue diet designed to normalize calcium and phosphorus, serum FGF23 levels increase dramatically suggesting that the effect of phosphate (and/or calcium) on serum FGF23 does not require vitamin D. Others have also documented an increase in serum FGF23 concentrations in mice with dose-dependent increases in phosphate ingestion .
Related posts:
 Osteoclast biology
Osteoclast biology
 Impact of physical characteristics and lifestyle factors on bone density and fractures
Impact of physical characteristics and lifestyle factors on bone density and fractures
 Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
 Relationship between periodontal disease, tooth loss, and osteoporosis
Relationship between periodontal disease, tooth loss, and osteoporosis
 On the evolution and contemporary roles of bone remodeling
On the evolution and contemporary roles of bone remodeling
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


