Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .


Introduction
Pheochromocytomas (PCCs) are rare neuroendocrine tumors that arise from the adrenal medulla and typically secrete catecholamines. When these tumors arise from the extraadrenal sympathetic or parasympathetic ganglia, they are called paragangliomas (PGLs). Pheochromocytoma (PCC) and paraganglioma (PGL) are together referred to as PPGLs, as they belong to the same family of neural crest-derived neoplasms. The estimated prevalence of PCC among patients with hypertension attending general outpatient clinics is low (0.1%–0.6%) [ ]. The annual incidence is approximately 2–8 per million [ ] in the general population. However, PCC is found in 5%–7% of patients with incidentally discovered adrenal masses [ ], which justifies the need to rule out PCC in patients with incidentally discovered adrenal tumors [ ].
Although the classical clinical picture of episodic hypertension associated with postural hypotension, palpitations, sweating, and headache may suggest the possibility of a PCC, this type of presentation is rather uncommon [ ]. The nature of the genetic mechanisms involved in the tumorigenesis, pathophysiology, molecular aspects, and the clinical profile of PCCs are complex. These often pose significant challenges to clinicians and laboratory scientists in the diagnostic workup, genetic evaluation, and the development of management algorithms, and in planning follow-up for these uncommon endocrine tumors. This chapter elucidates the pathobiological aspects, diagnostic evaluation, and management of PCCs and endocrine hypertension with the most up-to-date evidence.
Molecular and genetic aspects of PCCs
Understanding the genetic abnormalities and the consequent molecular alterations of PCC is essential for adequate diagnostic workup, treatment, and surveillance of patients and their families [ ]. Up to 70% of PPGLs are genetically determined by either germline (up to 40%) or somatic (30%–40%) mutations in one of the known susceptibility genes [ , ]. Mutations in the von Hippel Lindau ( VHL ), RET , and Neurofibromin 1 ( NF1 ) genes occur predominately in PCCs
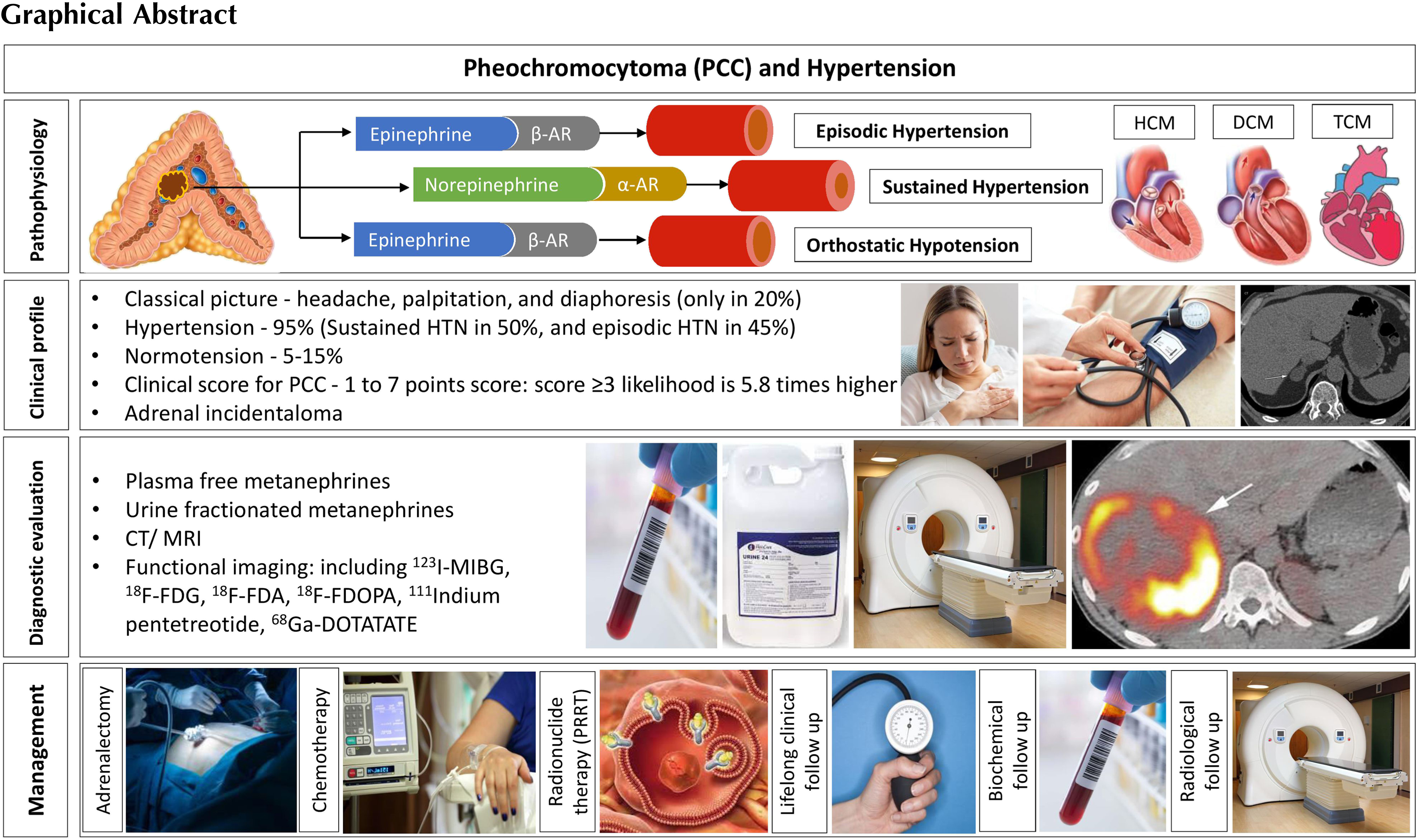
Tumor clusters
Cluster 1 tumors are defined by a pseudohypoxic molecular signature and accelerated angiogenesis. Pseudohypoxic PPGL can be further classified into Krebs cycle-related PPGL (Cluster 1A) and VHL/Endothelial PAS domain-containing protein 1 ( VHL/EPAS1 ) related PPGL (Cluster 1B). The cluster 1A tumors are associated with mutations in succinate dehydrogenase subunits ( SDH x or SDHA/SDHB/SDHC/SDHD ), succinate dehydrogenase complex assembly factor-2 ( SDHAF2 ), fumarate hydratase ( FH ), malate dehydrogenase 2 ( MDH2 ), glutamic-oxaloacetic transaminase ( GOT2 ), 2-oxoglutarate-malate carrier (SLC25A11), dihydrolipoamide S-succinyltransferase ( DLST ), and isocitrate dehydrogenase 1 (IDH1) . In contrast, the cluster 1B tumors are associated with mutations in VHL , hypoxia-induced factor 2α ( HIF2A ), Egl-9 prolyl hydroxylase-1 and -2 (EGLN1/2 encoding PHD1/2 ) , and iron regulatory protein 1 (IRP1) [ ]. The pseudohypoxic molecular signature is partially explained by the capacity of SDHx (cluster 1A) and VHL (cluster 1B) mutated cells to enhance the expression and decrease the degradation of hypoxia-inducible factors (HIFs). Moreover, an important consequence of SDHx inactivation is the accumulation of succinate and consequent chronic hypoxic stimulation. Thus, in summary, activation of the hypoxic pathway in VHL and SDH -related PPGLs results in enhanced expression and stabilization of HIFs (especially HIF2α), which in turn regulate/promote angiogenesis, energy metabolism, tumor progression, migration, invasion, and metastasis [ ].
The signature of the second cluster involves the upregulation of kinase signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway, the Ras/Raf/mitogen-activated protein kinase/extracellular-signal-regulated (ERK) kinase (MEK)/ERK cascade, and the mammalian target of rapamycin (mTOR) pathway, promoting cell growth and survival. These tumors develop due to gain-of-function mutations in RET or mutations in genes such as NF1, transmembrane protein 127 (TMEM127), myc-associated factor X (MAX), MET receptor tyrosine kinase (MET), MER Proto-Oncogene Tyrosine Kinase (MERTK) , Fibroblast Growth Factor Receptor 1 ( FGFR1) , and Harvey rat sarcoma viral oncogene homolog ( HRAS) [ ]. The third cluster is less explored, comprising genes leading to activation of the Wnt/beta-catenin pathway resulting in increased angiogenesis, cell proliferation, and invasion. Sporadic PPGLs with somatic variants in cold shock domain-containing E1 ( CSDE1 ) and mastermind-like transcriptional coactivator 3 ( MAML3 ) fusion genes were also reported in this cluster.
Clinical and biochemical characteristics of PCC: genotype-phenotype correlations
The catecholamines epinephrine and norepinephrine are synthesized by chromaffin cells in the adrenal medulla in response to acetylcholine (ACh) released by the preganglionic neurons of the sympathetic system. Most hormonally active PCCs produce a combination of norepinephrine and epinephrine, some cases exclusively norepinephrine (noradrenergic phenotype), and a much smaller proportion exclusively epinephrine. These differences in catecholamine production reflect differences in the expression of phenylethanolamine-N-methyltransferase (PNMT), the enzyme in the catecholamine biosynthetic cascade that converts norepinephrine to epinephrine [ , ].
The biochemical phenotype of PCCs can indicate specific molecular alterations [ ]. PCCs in MEN2 and NF1 patients express PNMT and secrete epinephrine, with an increase in plasma or urine metanephrines. In contrast, those PCCs related to VHL syndrome do not express PNMT and consequently do not produce epinephrine, but produce norepinephrine, as reflected by the solitary increases in plasma or urine levels of normetanephrines [ , ]. Similarly, tumors associated with the SDHx syndromes also do not express PNMT or produce epinephrine. They are characterized by norepinephrine production, but can also produce dopamine, manifested by increased normetanephrine and 3-methoxytyramine (3MT) levels [ ]. A dopaminergic phenotype (with increased 3MT levels) suggests a deficiency in the enzyme dopamine-beta-hydroxylase, which converts dopamine to norepinephrine. Tumors that produce predominantly or exclusively dopamine are rare and are usually found as extra-adrenal paragangliomas [ ]. These patients tend to be normotensive, which poses a substantial diagnostic challenge.
Genotype-phenotype correlations might impact the disease presentation, including catecholamine production. They have important implications for how genetic testing may be directed, or the results of testing interpreted, mainly if the conventional molecular testing is performed. When a PCC clinical presentation strongly suggests mutation in a specific gene (e.g., RET or VHL ), then targeted genetic testing for a germline mutation might still be appropriate [ ].
PCCs diagnosed at young ages suggest the presence of a germline pathogenic variant. Large tumors at diagnosis, extra-adrenal localization, and noradrenergic phenotype point toward germline SDHx gene variants (cluster 1), while adrenal localization and adrenergic phenotype point toward cluster 2 genetic abnormalities [ ]. Moreover, cluster 2 tumors exhibit high symptom scores (see below) and episodic symptoms that indicate epinephrine secretion. Cluster 1 tumors and probably cluster 3 tumors exhibit aggressive phenotype with higher metastatic risk [ ] ( Table 11.1 ).
| Mutation | Association with PCC | Metastatic risk | Biochemical phenotype | Cluster |
|---|---|---|---|---|
| RET | Almost exclusively located adrenally (unilateral or bilateral) | <5% | Adrenergic | Cluster 2 |
| NF1 | Almost exclusively located adrenally (often unilateral) | 2%–12% | Adrenergic | Cluster 2 |
| TMEM127 | Almost exclusively located adrenally (unilateral or bilateral) | Low | Adrenergic | Cluster 2 |
| MAX | Almost exclusively located adrenally (bilateral) | 10% | Adrenergic and noradrenergic | Cluster 2 |
| SDHB | Less common (often unilateral) | 35%–75% | Noradrenergic and/or dopaminergic | Cluster 1 |
| SDHA | Very rare | 30%–66% | Cluster 1 | |
| SDHC | Less common | Low | Noradrenergic | Cluster 1 |
| SDHD | Less common (unilateral or bilateral) | 15%–29% | Noradrenergic and/or dopaminergic | Cluster 1 |
| HIF2A/EPAS1 | Common | >30% | Noradrenergic | Cluster 1 |
| VHL | Very common (unilateral or bilateral) | 5%–8% | Cluster 1 |
Pathophysiology
Catecholamines (dopamine, norepinephrine, and epinephrine) are synthesized in and secreted from the chromaffin cells in the adrenal medulla and the sympathetic ganglia. The PNMT is not expressed by the sympathetic ganglia, meaning that most of the epinephrine in circulation is produced by the adrenal glands. Catecholamines act through the G-protein coupled adrenoceptors to regulate broad physiological processes [ ]. Norepinephrine signals through the α 1 , α 2 , and β 1 adrenoceptors, whereas epinephrine primarily stimulates the β 1 and β 2 adrenoceptors. Dopamine, on the other hand, has minimal effect on the adrenoceptors at physiological concentrations, but can stimulate both α and β adrenoceptors if the levels are increased as in dopamine secreting PPGL, leading to vasoconstriction and increased heart rate [ ].
α 1 -adrenoceptors are found on peripheral arteries and veins, causing vasoconstriction upon stimulation. Their stimulation increases the systemic blood pressure and reduces organ perfusion. It also induces positive inotropic effects in the cardiomyocytes. Stimulation of the α 2 -adrenoceptors on smooth muscle cells results in peripheral arterial vasodilation and coronary vasoconstriction. β 1 -adrenoceptors are stimulated by both norepinephrine and epinephrine and have a positive inotropic effect in the cardiomyocytes. β 2 -adrenergic receptors respond mainly to epinephrine and induce vasodilation of muscular arteries [ ]. Therefore, the concentration and type of catecholamine define the cardiovascular response. Acutely, catecholamines increase the heart rate, vascular resistance, and myocardial contractility. However, sustained catecholamine excess states would result in desensitization of the adrenoceptors. Several patients with PCC-related catecholamine excess are asymptomatic, and studies on the human and animal models report significant desensitization of both the α and β adrenoceptors [ ].
Clinically, several cardiovascular complications arise from excessive catecholamine production. Catecholamine excess states can lead to an acute increase in arterial stiffness and tachyarrhythmias. Chronic catecholamine excess states can cause myocardial ischemia and hypertension. PCCs can also contribute to the development of chronic (hypertrophic, dilated, obstructive), or acute (ischemic, takotsubo) cardiomyopathies [ ]. Moreover, catecholamine overproduction can lead to hypertensive encephalopathy, while paroxysmal hypertension can cause hemorrhagic or ischemic stroke. Vasoconstriction can also lead to renal failure as well as muscle ischemia and consequent rhabdomyolysis [ ]. For further reading on catecholamines and blood pressure regulation, please refer to Chapter 2 .
Metastatic risk
All PCCs have the potential to metastasize. Since no reliable biomarker has yet been identified that can accurately predict the metastatic potential of the disease, PCC-related malignancy is defined by the presence of distant metastasis in places where no chromaffin cells are physiologically found. Therefore, the term “malignant” has been replaced by “metastatic” PCC [ , ]. The presence of SDHx mutations, tumor size greater than 5 cm, multifocality, and the noradrenergic or dopaminergic biochemical phenotypes are the common risk factors for the development of metastatic disease [ ].
Several relevant biomarkers have been suggested as surrogates of the metastatic potential of PCCs and can contribute to generating various grading scores, such as the Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) grading system, the Grading of Adrenal Pheochromocytoma and Paraganglioma (GAPP), and the Composite Pheochromocytoma/paraganglioma Prognostic Score (COPPS) [ ].
Both PASS and GAPP scoring systems have been used globally to evaluate the malignant potential of patients with PCCs. PASS score of four or more has a high sensitivity for malignant behavior (sensitivity of 100% and specificity of 75%) [ ]. A tumor with GAPP score of less than 3 has a metastatic rate of 3% and 5-year survival of 100% [ ]. This reflects the ability of both scores to “rule out” the metastatic or malignant behavior of PCCs.
Clinical presentation
The classic triad of symptoms associated with PCCs is headache, palpitation, and diaphoresis [ ], which occur with a much higher frequency than other symptoms [ , ]. However, the complete triad is only found in about 20% of patients [ ]. The most common sign associated with the disease is hypertension, found in up to 95% of patients [ ]. The clinical presentation of hypertension can be (i) sustained hypertension, found in about 50% of patients, (ii) paroxysmal hypertension, found in 45% of patients, and (iii) normotension in about 5%–15% of patients [ , ]. Recently, based on the clinical signs and symptoms, a scoring system has been proposed ( Table 11.2 ) in order to establish the pretest likelihood of the disease (symptom score ≥ or < 3). Higher scores were associated with higher biochemical indices of catecholamine excess, but not with tumor size [ ]. Cluster 2 tumors are more likely to be related to episodic “flares” and paroxysmal symptoms, while tumors related to cluster 1 tend to present with sustained hypertension. Cluster 2 patients are more likely to present with pallor, tremor, and anxiety or panic attacks. Notably, clinically advanced PCC and PGL are now recognized as the commonest nonthyroid endocrine malignancies [ ].
| Clinical score for the likelihood of PCC b , c | Point score |
|---|---|
| Pallor | +1 point |
| Palpitations | +1 point |
| Heart rate ≥85 bpm | +1 point |
| Tremor | +1 point |
| Nausea | +1 point |
| Body mass index (BMI) > 30 kg/m 2 ) | −1 point |
| BMI < 25 kg/m 2 | +1 point |
a Adapted from From Geroula A, Deutschbein T, Langton K, Masjkur J, Pamporaki C, Peitzsch M, Fliedner S, Timmers HJLM, Bornstein SR, Beuschlein F, Stell A, Januszewicz A, Prejbisz A, Fassnacht M, Lenders JWM, Eisenhofer G. Pheochromocytoma and paraganglioma: clinical feature-based disease probability in relation to catecholamine biochemistry and reason for disease suspicion. Eur J Endocrinol. 2019;181(4):409–20. https://doi.org/10.1530/EJE-19-0159 .
b Cluster 1 tumors usually are associated with lower scores but sustained hypertension.
c Cluster 2 tumors usually are associated with higher scores and episodic “flares”.
Diagnostic workup
Biochemical testing
Initial evaluation of suspected PCCs should be performed with plasma free metanephrines or urinary fractionated metanephrines, as per the current clinical guidelines [ , ]. Considering appropriate preanalytical precautions (e.g., at least 20 min of supine rest, avoidance of potentially interfering medications sufficiently prior to testing), plasma free and urine fractionated metanephrines have a high diagnostic sensitivity for PCC/PGL. There is no clear benefit of measuring metabolites in plasma as opposed to urine, as both have excellent negative predictive values (>99%) at similar specificities (∼94%) [ , ]. Thus, a negative plasma free metanephrine or urinary fractionated metanephrine test virtually rules out a PCC/PGL except in those with very small tumors or in those with nonsecretory head and neck PGLs [ ].
The plasma free metanephrines have an equivalent diagnostic accuracy to urinary fractionated metanephrines in those with low risk for PCC/PGL as in those with symptoms suggestive of PCC/PGL. However, plasma free metanephrines have a superior diagnostic accuracy in those with high risk for PCC/PGL as in those with adrenal incidentaloma or in those on surveillance [ ]. The analytic method of choice is liquid chromatography-tandem mass spectrometry (LC-MS/MS), as it provides greater diagnostic performance when compared to alternative detection methods in the form of superior accuracy, cost-effectiveness and minimal analytical interferences with the drugs [ ].
Detection of dopamine-producing tumors or biochemically silent PCC/PGL remains a challenge, as these tumors rarely produce excessive amounts of adrenaline and noradrenaline. Therefore, a personalized approach for biochemical screening must consider phenotype-genotype-biochemical correlations. Upon suspicion of this type of tumor (specially SDHx -related tumors), the inclusion of plasma 3-methoxytyramine (3MT; the dopamine metabolite) measurement has been proposed and endorsed by the European Society of Endocrinology clinical practice guideline [ ], as well as by the European Society of Hypertension working group on endocrine hypertension position statements [ ], as a tool to increase the diagnostic sensitivity [ , ].
In a recent research study that evaluated adrenal incidentalomas, an approach that targeted plasma metabolomics (including 19 steroids, normetanephrine, and metanephrine) combined with adrenal lesion size provides 95% sensitivity and 99% specificity for PCC diagnosis. Nevertheless, discrimination of PCC from other adrenal secreting tumors is utterly dependent on plasma free metanephrines [ ]. Chromogranin A (CgA) can be a valuable plasma biomarker for patients with SDHB -related PCCs and sympathetic paragangliomas with normal plasma/urine metanephrines [ ].
The personalized interpretation of the test report is essential since conditions associated with blood sampling (e.g., supine, at rest), as well as chronic kidney disease, might alter the test results [ ]. Higher upper cut-off values are needed in patients with chronic kidney disease [ ]. Several drugs may cause falsely elevated results for the plasma or urinary metanephrines, both due to pharmacodynamic interference (e.g., antidepressants, sympathomimetics, MAO-inhibitors, levodopa, phenoxybenzamine) or analytical interference (e.g., acetaminophen, sotalol) (minimal for LC-MS/MS). A list of the most common interfering drugs can be found in the European and Endocrine Society guidelines [ , ].
Conditions with chronic sympathetic overactivity, including heart failure and obstructive sleep apnea, inappropriate sampling, drug interferences, and incorrect cut-off values can cause false-positive metanephrines test tests [ ]. Food items interfere with the 3MT estimation, hence overnight fasting and avoidance of certain foods are relevant for 3MT analysis [ ]. Solitary elevation of metanephrines/normetanephrines by 2-fold or simultaneous elevation of two or more metabolites (metanephrines, normetanephrines, and/or 3MT) are less likely to be false-positive [ ]. These situations need further diagnostic imaging, regardless of the pretest probability. On the other hand, in those with less than 2-fold elevation of a single metabolite with no other possible explanation for a false-positive test result, especially in those with a high pretest probability for PCC/PGL based on the clinical score given in Table 11.2 , a clonidine suppression test can be done to differentiate the false-positive test from the true-positive. However, those with a low pretest probability for PCC/PGL do not need further tests [ , ].
Diagnostic imaging
Anatomical imaging
Ideally, imaging studies should be carried out only after biochemical confirmation of the disease. Exceptions for this rule might be patients critically ill and/or in the intensive care setting [ ], as well in those where nonsecretory tumors are suspected. Appearance of PCC in the anatomical imaging is highly variable, as tumors can contain necrosis, hemorrhage, cystic changes, and calcifications. Upon suspicion of PCC, a contrast-enhanced computed tomography (CT) is the first-choice imaging modality because of its adequate spatial resolution and high sensitivity [ , ]. The demonstration of an adrenal tumor with an unenhanced CT attenuation value ≤ 10 Hounsfield Units (HU) has a high diagnostic predictive value to exclude the presence of PCC (sensitivity of >99%) [ , ]. Magnetic resonance imaging (MRI) also has good sensitivity and specificity for the diagnosis of PCC. It is the method of choice in patients allergic to iodinated contrast agents, and in those for whom radiation exposure should be limited (i.e., children, and pregnant women). A high intensity (bright) T2-weighted signal on MRI is highly specific for PCC, but it lacks sensitivity, as it occurs in less than half of the tumors [ ].
Functional imaging
The choice of the most appropriate method depends on each patient’s genotypical and/or phenotypical findings. Molecular imaging tracers for the diagnosis of PPGL can be classified according to their target ligand ( Table 11.3 ). Functional imaging modalities can benefit patients at high risk for metastatic disease, including those with large tumors, extra-adrenal tumors, and multifocal disease.
| Target ligand | Tracer | Indication | Comments |
|---|---|---|---|
| Catecholamine storage and synthesis | 123 I-MIBG | Metastatic PCC when radiotherapy with 131 I-MIBG is planned | Uptake by the normal adrenal gland. Lower accuracy for SDHx -related PPGLs |
| 18 F-FDA | Primary PCCs | Unavailable in most centers | |
| 18 F-FDOPA | Cluster 1B and 2 | Reduced sensitivity for SDHx -related PPGLs. Lack of uptake by healthy adrenal tissue | |
| Glucose metabolism | 18 F-FDG | Scanning patients with metastatic disease | Better than 123 I-MIBG for scanning patients with metastatic disease. Complementary with other functional imaging studies. |
| Somatostatin receptor | 111 Indium-pentetreotide (OctreoscanTM) | Complementary investigation | Provides complementary information to 123 I-MIBG for detection of metastatic PCCs |
| 68 Ga-DOTATATE | Diagnosis staging; considered for evaluation of metastases and multifocality; planning PRRT | High sensitivity for PCC with unknown genetic status |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


