Fig. 7.1
A 23-year-old female who presented with severe left flank pain with normal blood pressure. Biochemical testing revealed elevated plasma and urine normetanephrine levels. Anatomic imaging showed the following results. (a) A large right suprarenal mass (thick arrow) measuring 8.5 × 6.3 × 7.2 cm. (b) The left adrenal gland is normal in size (thin arrow). (c) Axial images show the mass abutting the right lobe of the liver with heterogeneous enhancement on contrast. (d) T2-weighted MRI shows a heterogeneously enhancing right suprarenal mass. Patient had right adrenalectomy and the tumor was confirmed to be pheochromocytoma on pathology
Desensitization of β-adrenoceptors can occur with prolonged stimulation of cells leading to diminished responsiveness of the cells to subsequent activation by catecholamines [53]. Other substances known to contribute to the clinical picture of pheochromocytoma include vasoactive intestinal peptide (VIP), ACTH, neuropeptide Y (NYP), atrial natriuretic peptide (ANP), growth hormone-releasing factor, somatostatin, parathyroid hormone-related peptide, calcitonin, and serotonin [54].
Sudden massive catecholamine release can cause severe vasoconstriction that can lead to severe cardiac dysrhythmias and pulmonary edema [55]. This can reflect on electrocardiography as prolonged QT interval and ST-T wave abnormalities mimicking coronary artery disease [56, 57]. Cardiomyopathy, acute myocarditis, cardiogenic shock, and sudden death are rare yet lethal complications that have been associated with pheochromocytoma [57–59]. Although the cause may be multifactorial and complex, injury to the myocardium from excess catecholamine plays a significant role. Pathologic examinations have revealed multifocal myocyte necrosis and replacement fibrosis with mixed inflammatory inflammation [60]. This can lead to intravascular hypovolemia and decreased cardiac output. The influence of catecholamines on vascular wall growth and thickening is believed to be the reason why there is a significant increase of intima-media thickness (IMT) in the common carotid artery of patients with pheochromocytoma [61].
Reversal of clinical manifestations and improvement in cardiac function were reported in several studies suggesting that early detection and intervention can prevent these fatal consequences [57–69, 62, 63].
Biochemical Evidence of Pheochromocytoma
Pheochromocytoma cells continuously metabolize catecholamines to metanephrines. Metanephrines reflect the actual levels of production of catecholamines in chromaffin cells. Subsequent studies have established that measurement of plasma or urine metanephrines for the diagnosis of pheochromocytoma was found to be superior to all other previous diagnostic tests [64–66]. Plasma metanephrines are measured in the free form while the urinary metanephrines are measured in the deconjugated form, both of which have advantages and disadvantages but with comparable results [66]. Measurement of plasma or urinary methoxytyramine may be done to detect DA-producing tumors [67]. Elevations of more than four times above the reference interval for both plasma and urine fractionated metanephrines are associated with almost 100% probability of having pheochromocytoma [64].
At present, liquid chromatography with tandem mass spectrometry (LC-MS/MS) is the ideal method for measuring plasma and urine fractionated metanephrines [68, 69]. This provides an effective method of purification with reduced interference from chromatographic contaminants such as medications. This method enables combined measurement of plasma NE, EPI, DA, and other metabolites such as DOPA, the precursor of catecholamines, and DOPAC, the deaminated metabolite of DA. While DHPG reflects the intraneuronal turnover of norepinephrine DOPA provides information on synthesis and transmission catecholamines. This enables comprehensive analysis of the dynamics of the patient’s catecholamine system. Its limitation is primarily the cost and availability as only specialized centers are equipped to run these methods. High-pressure liquid chromatography (HPLC) remains the most common analytical method for biogenic amine quantitation. Its overall sensitivity and specificity is reported at 98 and 92%, respectively [70]. Enzyme-linked immunosorbent assay (ELISA) may also be used to determine the levels of metanephrines at sensitivity lower than liquid chromatography [71].
The phenotype of hormone secreted by the tumor can also provide information about the size and location of the tumor [72]. There is a strong positive relationship between tumor diameter and summed plasma concentrations of free normetanephrine and metanephrine [72]. Adrenal tumors produce relatively high concentrations of plasma-free metanephrine compared to normetanephrine. Comparing sporadic and familial pheochromocytoma, biochemical parameters were found to be higher in sporadic than hereditary tumors [65]. Characteristic secretion patterns for syndromic pheochromocytomas reveal that MEN 2 patients secrete predominantly high metanephrines while VHL patients secrete more normetanephrine [18, 72] (Fig. 7.2).
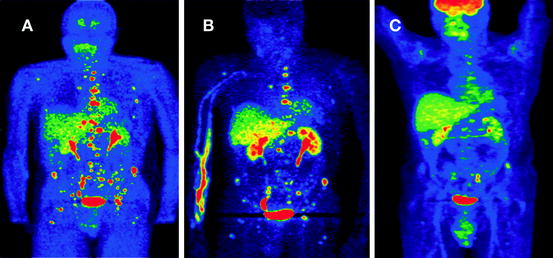
Fig. 7.2
A 24-year-old male presented with episodic hypertension. Several of his relatives were previously diagnosed with pheochromocytoma. Biochemical evaluation showed very high levels of plasma and urine normetanephrine. CT and MRI revealed a 3.6 × 2.6 × 2.8 cm retroperitoneal mass medial to the left kidney with multiple metastatic bone lesions. Functional imaging reveals the extent of metastatic disease using (a) FDA, (b) FDOPA, and (c) FDG PET imaging. Increased uptake of tracer is noted on the lumbar vertebrae, sacrum, and pelvis. The primary tumor was removed and the patient underwent chemotherapy. Genetic testing revealed that he had an SDHB mutation
Confounding Factors
Medications known to affect the results include phenoxybenzamine and tricyclic antidepressants, which are most frequently associated with false-positive results in plasma and urinary norepinephrine and normetanephrine. Selective α1-adrenoceptor blocking drugs, doxazosin, terazosin, and prazosin only increased urinary norepinephrine levels. β-Adrenoceptor blockers increase plasma metanephrine and urinary NE levels. Calcium channel blockers elevate plasma norepinephrine. Diuretics, angiotensin converting enzyme inhibitors, and angiotensin-1 receptor blockers have no influence on frequencies of false-positive biochemical results for any of the analytes [73].
Patient positioning at the time of blood extraction was found to affect the levels of plasma metanephrines. Upper reference limits of plasma-free metanephrines were higher in samples obtained from seated patients without preceding rest than from supine patients with preceding rest [74]. Catecholamine rich-diet only had substantial effects on plasma and urinary levels of free and deconjugated methoxytyramine [75].
An intravenous catheter should be inserted on the arm prior to letting the patient rest comfortable in the supine position for at least 20 min. Blood will be drawn from the catheter with the patient in supine position. Blood tests are best taken in the morning after an overnight fast.
Pharmacologic Test
The clonidine suppression test is utilized to distinguish between patients with pheochromocytoma and other causes of sympathetic activation [76]. Clonidine activates α2-adrenoceptors in the brain and sympathetic nervous system to suppress the release of NE. Baseline blood samples are taken for plasma normetanephrine followed by clonidine given at 0.3 mg/70 kg dose. Repeat plasma normetanephrine determination after 3 h should show a decrease in plasma normetanephrine of 40% from baseline values. Positive response consistent with pheochromocytoma is a lack of clonidine-induced suppression combined with a plasma normetanephrine concentration above the upper reference limit. Sensitivity of this test was reported at 96% with a specificity of 98–100% [73]. The use of glucagon stimulation test is no longer recommended because of the lack of sensitivity and risk for hypertensive complications. Glucagon-provoked increases in plasma catecholamines only revealed the presence of the tumor in less than 50% of affected patients [77].
Ancillary Laboratory Tests
The pro-hormone chromogranin A is also being utilized as an adjunct biochemical test in the diagnosis of pheochromocytoma [78–80]. It is secreted by a variety of peptide-producing endocrine neoplasms including pheochromocytoma, parathyroid adenoma, medullary thyroid cancer, carcinoids, oat-cell lung cancer, pancreatic islet-cell tumors, and aortic-body tumor [78]. Studies have established significant positive relationship between chromogranin A levels, tumor mass, and malignancy [81–83]. Serial measurements can be used to monitor treatment response.
Patients with pheochromocytomas may present with hyperglycemia because of α2-adrenergic inhibition of insulin release, EPI-induced inhibition of glucose uptake, adrenergic stimulation of hepatic glucose production, and α-adrenergic receptor desensitization. In young patients with hypertension and normal body weight, the presence of diabetes is a clinical clue to the diagnosis of pheochromocytoma [84, 85].
The main effect of the excess catecholamine secretion on lipid metabolism was increased lipolytic activity, lower serum triglyceride (TGL), and increased high density lipoprotein (HDL) cholesterol concentrations [86]. Treatment of pheochromocytoma with β-blockers may worsen the lipid profile of these patients by decreasing HDL and increasing TGL. In contrast, the selective α1-blocker increases HDL cholesterol and decreases TGL.
Patients suspected of having MEN 2 syndrome should be tested for intact parathyroid and serum calcium to rule out primary hyperparathyroidism. Serum calcitonin levels are elevated in patients with medullary thyroid cancer. Complete blood count may reveal transient leucocytosis during spells. Polycythemia from erythropoietin may be noted in VHL patients.
Most of the metabolic derangements noted during the diagnosis of pheochromocytoma are usually improved or reversed after the tumor is removed.
Tumor Localization Using Anatomical Imaging
Generally, imaging studies for pheochromocytoma follow biochemical diagnosis. This should be initiated if clinical evidence is strong and biochemical tests are positive. Patients with previous history of the tumors and hereditary predisposition are the exception [64]. Anatomical imaging is done to provide precise localization while functional imaging reveals nature, activity, and extent of the lesions. The CT and the MRI can detect tumors within the adrenal with a minimum size of 0.5–1 cm in diameter and extraadrenal lesions of at least 1–2 cm [87]. CT is fast and readily available in most medical centers. MRI has higher sensitivity, superior contrast resolution for extraadrenal lesions, and lacks ionizing radiation. It is the procedure of choice for children, pregnant women, and patients with allergies to contrast material.
Unenhanced CT provides density measurements of the lesions while succeeding delayed contrast studies quantify the washout characteristics and surrounding vascularity [88, 89]. The attenuation coefficient of lesions expressed as hounsfield units (HU) has been used to differentiate adrenal adenomas from tumors such as pheochromocytoma; values more than 10 HU supports the diagnosis of pheochromocytoma [90, 91]. Most sporadic pheochromocytomas are described as 2–3 cm in diameter with smaller lesion with homogenous appearance and larger lesions with inhomogenous appearance attributed to hemorrhage and necrosis [92]. On MRI, the T1-weighted sequences are equal to or higher than that of the liver, kidney, and muscle. T2-weighted MRI appearance is variable with smaller lesions appearing homogenous and larger lesions heterogeneous. Increasing heterogeneity correlated pathologically with increasing amounts of hemorrhage, necrosis, and fibrosis [93]. Pheochromocytomas enhance avidly with contrast and have a prolonged washout period. The addition of MR angiographic sequences is helpful in assessing the vascular supply of paragangliomas particularly in carotid body tumors [94]. In general, the majority of paragangliomas arise from the adrenal medulla. Of the 15% found outside of the adrenals, 46% are found in the renal hilum, 29% of the organ of Zuckerkandl, 10% of the thoracic paraspinal region, 10% in the bladder, and 2–4% of the head and neck region [95]. By order of significance, initial scan should be done on the abdomen and pelvis followed by the chest and neck.
Tumor Localization Using Functional Imaging
Functional imaging studies are done to prove that the tumor is pheochromocytoma and to reveal its extent. Radiolabeled ligands specific for catecholamine synthesis and metabolism as well as nonspecific ligands are utilized in nuclear imaging techniques to detect pheochromocytoma [96]. Metaiodobenzylguanidine (MIBG) is an analog of NE that is taken up by the human norepinephrine transporter system (hNET) into sympathomedullary cells and stored in the secretory granules via the VMAT [96]. MIBG has high affinity to hNET system but does not bind to adrenergic receptors nor metabolized [97]. 123Iodine (123I) is preferentially used over 131iodine (131I) as label because it allows better image quality, greater sensitivity, and reduced radiation exposure due to higher photon flow and shorter half-life [98]. Overall sensitivity in several series ranged between 75 and 89% [99, 100]. Sensitivity was consistently higher for adrenal and sporadic pheochromocytoma [100, 101].
Positron emission tomography (PET) imaging is a functional imaging technique based on detection of coincident photons originating from the annihilation of emitted positrons with electrons from surrounding tissues [102]. Biological substances labeled with positron emitters reveal lesions within minutes or hours after administration. It produces superior quality of images and subject patients to relatively low radiation exposure [97]. Combined anatomical and functional imaging has now been used to localize lesions with greater accuracy [102]. Complementing functional and anatomic imaging facilitates the distinction between pheochromocytoma and physiologic tracer accumulation in normal adrenal tissue [103].
Specific radioligands pass through the membrane hNET and target-specific steps in catecholamine metabolism. 11C EPI and 11C-hydroxyephedrine (11C-HED) are NE analogs that were the first positron-emitting tracers for the sympathetic nervous system. 18F-fluorodopamine (FDA) is an analog of DA that is taken in by both the plasma membrane and intracellular vesicular transporters in catecholamine secreting cells. Several studies have reported the utility of FDA imaging in the diagnosis pheochromocytoma [104–106]. It has comparable results with MIBG in detecting non-metastatic pheochromocytomas but higher sensitivity for metastatic lesions [107]. Established standard uptake values (SUV) for physiologic uptake of normal adrenal glands was <7.3 while pheochromocytoma-related uptake was >10.1 [103]. FDA results in lower radiation dose and faster procedure time. Its use is advocated for the evaluation of biochemically established adrenal, extraadrenal, and metastatic paragangliomas in localizing primary tumors and ruling out multifocal and metastatic lesions [108].
Nonspecific functional imaging labeled tracers utilize the amino acid transport system, glucose transporter, and somatostatin receptor. 18Fluoro-l-dihydroxyphenylalanine (F-DOPA) is transported into the cell by the large amino acid transporter 2 (LAT2), then decarboxylated to 18F-FDA, and then transported inside the vesicles through the VMAT. Pretreatment with carbidopa enhances the sensitivity of 18F-DOPA for adrenal and extraadrenal paragangliomas by increasing the tumor to background ratio of tracer uptake [109]. This imaging was also found to be superior to MIBG scintigraphy with an overall sensitivity of 90% and specificity of 93% [110, 111]. 18F-DOPA has the advantage of having little physiologic uptake in healthy adrenal tissues compared to previously imaging modalities [112]. Combining 18F-DOPA with CT or MRI imaging improves sensitivity to 93–100% [110, 111]. Sensitivity of 18F-DOPA for metastases associated with SDHB appears to be limited [109].
18Fluoro-2-deoxy-d-glucose (FDG) PET scanning reflects excessive glucose uptake by the glucose transporter-1 (GLUT-1) among cancer cells related to a shift to anaerobic glycolysis known as the “Warburg effect” [113]. Uptake was reflective of inherent metastatic tendency and aggressive behavior [114]. Sensitivity was highest for SDHB metastatic lesions reaching 100%, a finding that is linked directly to the tumor biology of SDHB lesions [115]. False-positive results have been reported with areas with infection and inflammation [114].
Somatostatin receptor scintigraphy is used to detect the presence of somatostatin receptors types 1, 2a, and 3 in neuroendocrine tumors [116]. 111-Indium labeled octreotide is a radiolabeled somatostatin analog specific to somatostatin receptor 2 used for diagnosis and therapy for neuroendocrine tumors [117]. It can detect metastatic pheochromocytoma and head and neck paragangliomas with a sensitivity of 89% [118, 119]. Physiologic uptake is observed mainly in the kidneys [120]. Infection and inflammation are known to produce false-positive results [121].
In summary, the 18F-FDA and 18F-DOPA are the ideal imaging procedures to localize the primary tumor and rule out metastasis. The 18F-DOPA is particularly useful in patients with head and neck paragangliomas [122]. 18F-FDG is the best imaging procedure for SDHB-related pheochromocytoma. 123I-MIBG and octreotide scan serves as an alternative imaging procedures that are also used to evaluate possible response to radiotherapy. Bone scintigraphy plays a role in staging of malignant pheochromocytoma especially with SDHB mutation [123] (Fig. 7.3).
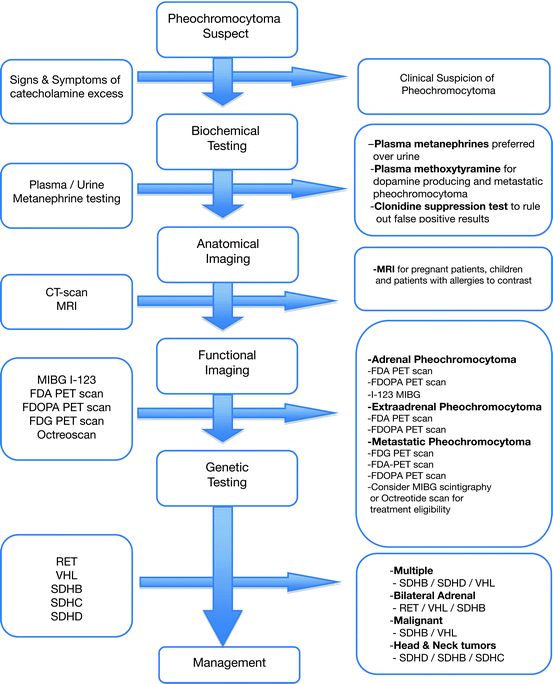
Fig. 7.3
Diagnostic approach to pheochromocytoma
Medical Treatment
Control of hypertension and accompanying symptoms are the immediate concerns that need to be addressed. The drug of choice is phenoxybenzamine, an irreversible, noncompetitive, α-adrenoceptor blocker. Its half-life is 24 h. Total daily dose of 1 mg/kg/day can be given in two to three divided doses and titrated accordingly. Initial dose can be started at 10 mg twice a day and increased in increments of 10–20 mg every 2–3 days [124]. Appropriate dosing can control paroxysms of hypertension and maintain BP at optimum level. Significant postural hypotension, reflex tachycardia, vomiting, and shock can be observed if the dose is too high.
Other α-blockers that can be used include prazosin given at 2–5 mg two to three times a day, Terazosin at 2–5 mg/day, and doxazosin at 2–8 mg/day. These are selective α1-adrenoceptor blockers that decrease total peripheral resistance and venous return. Severe hypotension may be observed after the first dose warranting it to be given at bedtime. Succeeding dose adjustments can be done more quickly with fewer side effects.
β-Adrenoceptor blockers compliment the action of α-blockers in controlling hypertension. They also help control tachycardia brought about by catecholamine release or induced by α-adrenoceptor blockers. β1-selective blocker atenolol can be given at 12.5–25 mg two to three times a day or Metoprolol given at 25–50 mg three to four times a day. Alternatively, nonselective beta adrenoceptor blockers such as propanolol, timolol, nadolol, penbutolol, and sotalol can be used. Combined alpha and β-adrenoceptor blockers are not recommended because the fixed ratio favors beta blocking over alpha blocking effect. Emphasis is placed on starting after starting α-adrenoceptor blockers before β-adrenoceptor blockers because unopposed α-receptor activation can result to hypertensive crises [125].
Calcium channel blockers can be used as the primary antihypertensive drug in low-risk patients [124]. As an alternative, it can replace adrenoceptor blockers in case side effects cannot be tolerated or when hypotension is noted on intermittently hypertensive pheochromocytoma patients since they do not cause hypotension during normotensive period [126]. These drugs block the norepinephrine-mediated calcium influx into vascular muscle controlling hypertension and tachyarrhythmias. It can also be used as an additional medication for patients who remain hypertensive despite maximal adrenergic blocker medications [124]. Amlodipine can be given at 10–20 mg/day, Nicardipine at 60–90 mg/day, Nifedipine 30–90 mg/day, and Verapamil at 180–540 mg/day.
Alpha-methyl-l-tyrosine or metyrosine (Demser) inhibits tyroxine hydroxylase, the rate-limiting step of catecholamine biosynthesis. It depletes catecholamine stores and contributes in the control of hypertension particularly in patients with biochemically active tumors [127–128]. Treatment can be started with 250 mg every 8–12 h and titrated up to 1.5–2 g/day 1–3 weeks before the surgery [124]. Side effects include fatigue diarrhea, anxiety, depression, and cystalluria [129]. Central effects can be explained by the ability of metyrosine to cross the blood brain barrier. Symptoms can resolve when doses are lowered or discontinued.
Preoperative Management
Adrenergic blockade can be started 1–2 weeks prior to scheduled surgery to allow ample time to control hypertension, control the heart rate, and expand contracted volume. Phenoxybenzamine can be titrated daily according to blood pressure response. β-Blocker can be added after 3 days for tachycardic patients. Metyrosine and a calcium channel blocker can be added in patients with uncontrolled hypertension [124]. Last doses of phenoxybenzamine and metyrosine are given at midnight prior to operation.
High salt diet is given a few days after starting α-adrenoceptor blockers to help restore diminished blood volume. Fluid replacement is initiated 2 days prior to surgery and continued throughout the procedure to expand the intravascular volume and reduces the frequency and severity of postoperative hypotension. Hypotension and shock can result from diffuse vasodilation when the tumor is removed.
Surgical Management and Approach
Laparoscopic surgery is the procedure of choice for most pheochromocytomas and sympathetic paragangliomas. It is safe and effective since blood loss was significantly less, postoperative recovery time was shorter, and the procedure is less invasive [130–134]. Option to convert to open surgery should be considered in cases with unexpected difficulty in dissection, invasion, or adhesions [134–138]. Long-term follow-up of post-laparoscopic adrenalectomy patients confirmed favorable outcome beyond 3 years after the procedure [139]. Bilateral cortical sparing surgery should be considered for familial cancer syndromes with bilateral or multiple tumors to avoid the lifelong consequences of bilateral adrenalectomy [140, 141].
These tumors show enormous variability in cytology and histologic pattern that must be differentiated from other neuroendocrine tumors. Gross examination can reveal multiple and bilateral tumors that suggest familial disease. Malignancy is defined as the presence of metastases; local invasion alone is a poor predictor of metastases and the absence of apparent invasion does not preclude the development of metastases [142]. There is no histologic feature nor scoring system that can predict metastatic potential. Standardized pathologic reporting is advocated by the United States Association of Directors of Anatomic and Surgical pathology (ADSP) and the United Kingdom Royal College of Pathologist to include the list of potentially unfavorable findings that can help identify potentially malignant tumors. Immunohistochemical staining for Ki-67 on paraffin sections of monoclonal antibody MIB-1 has been correlated with malignancy but available data needs further support [138, 143] (Fig. 7.4).
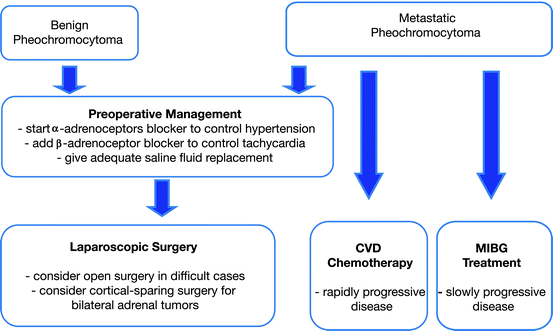
Fig. 7.4
Therapeutic approach to pheochromocytoma
Crisis Management
Considering the amount of hormones produced, it is likely that the stress of surgery can trigger the release of vast amounts of catecholamines resulting to uncontrolled hypertension and overall morbidity. Management requires the use of drugs that can be titrated with rapid action and short duration.
Sodium nitroprusside is a nonselective arterial and venous vasodilator that can be given as an infusion. It acts within 30 s and peaks within 2 min and loses its effect after 3 min. Hypertension can be controlled at doses between 0.5 and 5 μg/kg/min [144]. Doses >10 μg/kg/min for >10 min must be reduced or a second agent should be added because of risk of toxicity [145]. This is the first-line drug used to control intraoperative crisis.
Nicardipine is an intravenous dihydropyridine calcium channel blocker that can be utilized to prevent catecholamine-induced coronary vasospasm. Loading dose can be started at 1–2.5 mg for the first 2 min as bolus then maintained as an infusion at 5–15 mg/h [146].
Magnesium sulfate is beta agonist that acts as a vasodilator and antiarrhythmic. It can be used to control the manifestations of catecholamine excess by itself or in combination with other agents [147]. Loading dose of 1–2 g IV can be given, and then continued as IV infusion at 1–3 g/h.
Phentolamine is an intravenous competitive, nonselective α-adrenergic receptor antagonist, with a half-life of 19 min, and may be used as initial therapy for hypertensive crisis as an alternative if other medications are not available. A bolus of 5–15 mg can be given, and then infused at 12–120 mg/h.
Esmolol, a short acting selective β1-adrenergic adrenoceptor antagonist, can be used as adjuvant therapy for uncontrolled tachycardia. Loading dose of 500 μg/kg for 1 min then followed by an infusion at 50–300 μg/kg/min.
Hypotension can occur during surgery particularly after venous clamping and tumor isolation [148]. This can be managed by judicious administration of IV fluids or use of appropriate vasopressors. Phenylephrine can be given to cause α-adrenergic-mediated vasoconstriction without provoking catecholamine release. Alternatively, vasopressin may be used in small bolus doses and continued as infusion [149, 150].
Hypoglycemia can occur as a result of rebound release of insulin secretion after the removal of the tumor. This can be corrected with intravenous infusion of glucose [151]. Blood sugar should be monitored continuously since periods of hypoglycemia may persist for several days [152].
Operative outcome among patients with pheochromocytoma has vastly improved in recent years because of better perioperative management. Nonetheless, success is dependent on close monitoring and critical care by an expert multidisciplinary team.
Malignancy Implications and Management
Prevalence of malignancy has been reported to be between 5 and 26% [153]. SDHB mutation was found in 30–43% of malignant pheochromocytomas and paragangliomas [34, 35, 154]. The most common sites of metastases are the lymph nodes, bones, liver, and lungs. They can be present at the time of diagnosis or appear during recurrence. Overall, 5-year survival rate is between 40 and 74% [155, 156]. It is more common among extraadrenal pheochromocytomas [155, 157]. Malignant pheochromocytoma associated with SDHB mutation generally has a poorer prognosis [33, 154].
The signs and symptoms of catecholamine excess can be manifested in clinically functioning malignant pheochromocytoma [158]. DA can preferentially be secreted due to alterations of catecholamine synthesis in malignant pheochromocytoma cells [155]. Measurement of plasma-free methoxytyramine together with metanephrine and normetanephrine may be useful for identification of these tumors [155]. Markedly elevated chromogranin A levels correlate well with plasma metanephrines, size of tumor, and malignancy.
Anatomical imaging with both CT scan and MRI can identify metastatic and extraadrenal lesions above 1 cm in diameter [159]. Both imaging procedures are vital in the accurate localization of metastatic tumors. Functional imaging defines the extent of the disease and potential response to radiopharmaceuticals.
There is no effective cure for metastatic pheochromocytoma but surgery remains a viable option for tumor reduction if it is technically feasible [153]. Most treatments are palliative, directed at reducing the tumor burden, decreasing hormonal activity, and improving the quality of life [160].
Total adrenalectomy and resection of loco regional lymph node or complete excision of paragangliomas and removal of distant metastases are recommended [161]. Hepatic metastases can be treated with arterial embolization, chemoembolization, cryoembolization, radiofrequency ablation, or transcatheter arterial embolization in specialized centers [162, 164]. The risk increases with the size of the tumor but does not reliably predict malignancy in pheochromocytomas with local disease only [137].
I-131 MIBG therapy is useful only for slow to moderately progressive metastatic patients with lesions that take up this radionuclide. Cumulative results of I-131 MIBG treatment show partial tumor response of 25.3% and stable disease of 43.4% [165]. Higher MIBG doses have been associated with hematotoxicity. Response to somatostatin receptor therapy has failed to show significant improvement on biochemical and clinical response [126, 166]. Chemotherapy is considered among rapidly progressive metastatic disease as first-line treatment [167]. The most evaluated regimen uses Cyclophosphamide, Vincristine, and Dacarbazine (CVD) given for 2 days and cycled after 21 days. Toxicity was mild to moderate with remission period less than 2 years [167]. Recently, reported survival analysis of patients treated under this regimen did not reveal any significant improvement but provided effective palliation for patients with progressive and advanced pheochromocytoma.
Pheochromocytoma in the Young
The clinical presentation of pheochromocytoma in the young can be classified into two, a biochemically functioning tumor or an inactive incidental retroperitoneal mass [165]. Symptomatic tumors manifest similarly to adults with severe and sustained hypertension as the most striking symptom [168]. Biochemically inactive tumors can present as a mass that is often mistaken for the more common neuroblastoma [165]. Both tumors are derived from the neural crest cells and share the capability to secrete catecholamines but pheochromocytomas produce physiologically active hormones.
Most of the adrenal pheochromocytomas occur as unilateral lesions but a significant number present as bilateral lesions at the time of presentation or within a few years [168]. Up to 84% of multifocal tumors have genetic predisposition, with the most common defect is associated with VHL gene [36].
Biochemical confirmation of the tumor can be done with urine or plasma determination of metanephrine levels. The MRI is the preferred imaging modality for children. This can be complemented by functional information using 123I-MIBG imaging, octreoscan, or PET scan using FDA or FDA for general evaluation and FDG for metastatic evaluation.
Preoperative therapy with phenoxybenzamine is given at 20–50 mg/day in two to three divided doses titrated to control hypertension [169]. Additional β-adrenergic blocker atenolol may be warranted in patients with uncontrolled hypertension, tachycardia, and dysrhythmias. This can be given at 20–60 mg daily in two to three doses. Metyrosine given at 250 mg/day two to four times a day can also be added to control adrenergic symptoms of the patient. Calcium channel blockers are alternative drugs that can be used to control hypertension. Volume replacement should be limited to 10 mL/kg/h because of the risk of catecholamine-induced pulmonary edema [170].
Laparoscopic surgery minimizes catecholamine-induced hemodynamic changes during surgery and overall perioperative morbidity [166, 171]. Adrenal sparing surgery should be considered because of the significant morbidity associated with the need for lifelong steroid replacement [166, 167]. Nonetheless, close follow-up is needed to check for possible recurrence. Malignancy is higher among young pheochromocytoma patients, a series reported an incidence of 47% [165]. Identified risk factors for the development of malignant tumors include a tumor size of >6 cm, extraadrenal lesion, and sporadic occurrence. Overall survival rate is favorable primarily because of early detection and aggressive intervention.
Summary
We discussed pheochromocytoma and extraadrenal paragangliomas as functional tumors of the neuroendocrine system. Both can secrete excessive amounts of catecholamines. Signs and symptoms are variable, but generally include hypertension.
Plasma or urine metanephrines are reliable biochemical tests that are sensitive to detect patients with biochemically active tumors. CT scan and MRI are anatomic imaging tests that remain vital for accurate preoperative assessment. Functional imaging can be diagnostic and therapeutic; it enables detection of tumor extent and assesses possible response to therapy.
Focus on genetic studies for familial predisposition for early detection is now at the forefront of research. Identification of SDHB mutations warrants aggressive treatment because of tumor propensity for metastasis with poor prognosis.
Surgery remains the cornerstone of treatment with potential cure for benign tumors and palliative management. Long-term follow-up is necessary to detect recurrence and metastasis. Malignancy still holds poor prognosis as therapeutic options remain limited. Radiopharmaceutical and chemotherapy remain promising but no major breakthrough has been achieved. Palliative care and lesion-based treatment remain the standard.
Acknowledgments
This research was supported, in part, by the Intramural Research Program of the NIH, NICHD.
References
1.
Delellis RA, Lloyd RV, Heitz PU, Eng C. Pathology and genetics of tumors of the endocrine organs. Lyon: IARC Press; 2004.
2.
Tischler AS. Pheochromocytoma and extra-adrenal paraganglioma: updates. Arch Pathol Lab Med. 2008;132(8):1272–84.PubMed
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


