Renal epithelial neoplasms are not a rare disease, having an incidence of approximately 10 cases per 100,000 population. Approximately 57,760 new cases of renal cortical and renal pelvic tumors will be diagnosed and 12,900 patients will die of the disease (1,2,3) (Table 39A.1).
Traditionally, renal tumors have been described by their cytoplasmic features (clear cell, granular, oncocytic, spindled, and rhabdoid) as well as their growth pattern characteristics (acinar, papillary, tubular, and sarcomatoid). While it is certainly true that virtually all renal cell carcinomas contain one or more of these cell types and growth patterns, a classification based solely on these distinctions is not very precise (Table 39A.2). For example, “sarcomatoid carcinoma” does not exist as an entity since we now know that sarcomatous features may be present not only in clear cell carcinomas but also in other types such as collecting duct, chromophobe, and papillary RCC. “Granular cell” features may be observed in tubulo-papillary, oncocytic, and collecting duct tumors, as well as high-grade clear cell carcinomas, all of which have very different natural histories. In 1986, Dr. W. Thoenes described a renal tumor which he called chromophobe renal cell carcinoma (4). During the ensuing years, he and his collaborators proceeded to reevaluate the classification of renal tumors based on morphologic, histochemical, immunohistochemical, and electron microscopic data (5). At the same time, advances in laboratory methodology allowed us, for the first time, to establish cell cultures and perform karyotypes on solid tumors, including RCC. During the interim, many investigators worked on the molecular characterization of renal tumors. We should not underestimate the contributions made to our understanding of the genetic basis of renal epithelial neoplasms by our colleagues studying hereditary tumors, particularly the group from the National Institutes of Health led by Drs. Zbar, Linehan, and Merino. Their work, combined with work done on sporadic tumors by others, has translated into a new morphologic classification that has been, at least in part, validated by genetics and molecular genetics (6,7,8,9) (Table 39A.1). Interestingly, the findings made by the morphologists and basic scientists seemed to validate each other, leading to a revised classification with greater scientific merit. More recently, clinical studies have provided further validity to the results (10).
TABLE 39A.1 EPITHELIAL TUMORS OF THE KIDNEY (PARTIAL LIST)
Several systems over the years have been proposed for the grading of renal tumors. Of these, the one proposed by Fuhrman and colleagues is the most widely used (11,12,13,14). This system uses nuclear grades based on nuclear size, irregularity of the nuclear membrane, and nucleolar prominence. Although we and others have demonstrated good correlation between Fuhrman nuclear grade and survival in clear cell (conventional) carcinomas, application may be cumbersome because it requires the pathologist to measure nuclear size. Although many of us believe that a different grading system with a better discriminatory power is necessary, we recommend that the Fuhrman’s grading system be used until a more reproducible and clinically relevant classification emerges. The utility of the Fuhrman’s or any other grading scheme in papillary and chromophobe cell carcinomas remains controversial. In papillary carcinoma, we and others have found it not to have clinical significance on multivariate analysis (15,16,17,18). Fuhrman nuclear grading appears to be inappropriate for chromophobe renal cell carcinoma, as it very frequently shows marked nuclear irregularities (grade 3 nuclei) in spite of the overall excellent survivals. There is no reason to grade renal oncocytomas since they are benign tumors. Fuhrman nuclear grade has never been applied to other variants of renal carcinoma such as collecting duct, medullary, or unclassified tumors.
In 1969, Robson and colleagues published a staging scheme that was adopted by most clinicians and pathologists and has been shown to correlate well with survival. During the ensuing years, the American Joint Committee on Cancer in collaboration with the Union Internationale Contre le Cancer published the TMN classification, the latest of which was released in 2009 (19,20). Undoubtedly, any of these classifications offer valuable prognostic information, but only within specific tumor types. For example, large size and extracapsular extension does not have the same prognostic significance in cases of oncocytoma or even chromophobe carcinomas that it has in clear cell (conventional) tumors. Up to 20% of renal oncocytomas may present with pT3 disease even though they are a benign tumor. A major improvement of the latest TMN classification is the recognition of the importance of the renal sinus, not only renal sinus fat invasion but also involvement of the muscular branches of the renal vein in this site, both of which are now considered pT3 disease (20). Recently, a series of publications have highlighted the importance of the renal sinus and sinus veins in the staging of RCC (21,22). Careful gross examination and adequate sampling of the renal sinus-tumor interface, especially in larger tumors, is strongly recommended.
IMMUNOHISTOCHEMISTRY OF RENAL EPITHELIAL TUMORS
Immunohistochemistry has the potential for use to differentiate between different subtypes of primary renal epithelial neoplasms, or to confirm the renal origin in a metastatic tumor. Numerous antibodies have been utilized and reported to be of use in both these situations (23,24,25,26,27) (Table 39A.3). In our experience, many of these antibodies are able to discriminate between better-differentiated tumor subtypes. However, such tumors are also easily classifiable on light microscopic evaluation. In situations where the antibodies are most often required, that is, in high-grade or poorly differentiated tumors, the results tend to be inconsistent and unreliable. Among the more recent antibodies, carbonic anhydrase IX (CAIX) appears to be the most useful in the differential diagnosis of renal cell tumors (27,28,29). It shows diffuse and strong membranous reactivity in more than 90% of clear cell RCC, compared to absent or at the most focal, usually perinecrotic positivity in other common subtypes of renal cell tumors. Diffuse reactivity is retained even in the sarcomatoid components of most cases of clear cell RCC. Other antibodies that we have found of practical utility in the differential diagnosis of renal cell tumors include c-kit (CD117), which consistently stains chromophobe RCC and renal oncocytoma in a diffuse manner, whereas most other tumors, even those with eosinophilic cytoplasm, are either negative or only focally positive. We also find diffuse positivity for CK7, as in a large proportion of cases of chromophobe RCC, quite useful in their distinction from renal oncocytoma.
IN VIVO DIAGNOSTICS IN THE EVALUATION OF RENAL TUMORS
Depending on the subtype and the stage of the renal tumor, as well as its expected biological behavior, clinical comorbidities, etc., multiple therapeutic options tailored to an individual patient are now being offered. Among others, such options include in situ ablation of the tumor, targeted therapies in a neoadjuvant setting, or even watchful waiting in selected cases. In view of these developments, availability of a robust and dependable panel of immunohistochemical stains has become even more important, as pathologists are frequently asked to render diagnosis on limited material. Using a select panel of five antibodies—CAIX, CK7, CD117, racemase, and CD10—we find a more than 90% diagnostic accuracy on ex vivo needle core biopsies of renal cell tumors (Table 39A.3). Immunohistochemical results are particularly useful if both qualitative and quantitative characteristics are taken into consideration (27). These numbers are similar to what is being reported in the literature lately (30,31,32,33,34). This fact indicates that if pathologists are provided with adequate biopsy samples, close attention to cytomorphologic features, together with a judicious use of immunohistochemistry, will allow for an accurate diagnosis in the overwhelming majority of the cases. The specimen adequacy rate in both ex vivo and in vivo series is roughly 80%, with most inadequate samples due to necrosis, sparse cellularity, or cystic change.
SPORADIC RENAL CELL TUMORS
Clear Cell (Conventional) Renal Cell Carcinoma
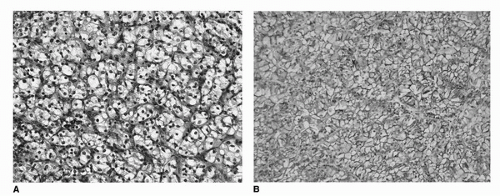
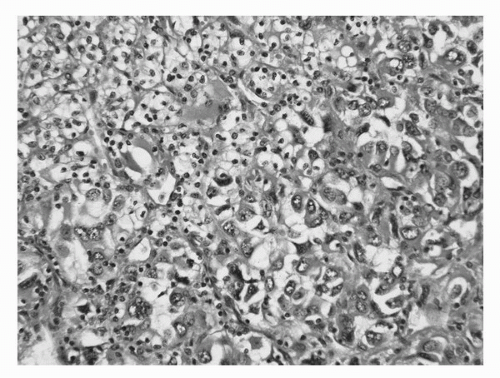
Conventional (clear cell) carcinoma comprises approximately 60% to 65% of all RCC (35). They are characterized by tumor cells with clear cytoplasm (due to increased lipid and glycogen) and an acinar growth pattern (Fig. 39A.1A,B). The “clearing” of the cytoplasm extends from the nucleus to the cytoplasmic membrane, especially in low-grade tumors. In higher-grade lesions, the cytoplasm may be only partially clear or mostly “granular,” that is, to say eosinophilic (Fig. 39A.2). It is in these areas where one is likely to see a loss of the acinar growth pattern, the tumor now being either solid or sarcomatous. It is important to look for areas of transition to a lower grade to establish the correct diagnosis. These tumors may take on a papillary or pseudopapillary appearance focally, but this is due to degenerative changes rather than true papillae formation.
TABLE 39A.3 IMMUNOHISTOCHEMICAL STAINING IN THE DIFFERENTIAL DIAGNOSIS OF RENAL EPITHELIAL TUMORS
EMA
CK Wide Spectrum
Vimentin
CAIX
CK7
AMACR
CD-117
TFE-3
TFE-B
HMB-45
A-103
PAX-8
CD-10
Clear Cell RCC
+
+ May be focal or patchy
+ Less apparent in low-grade tumors
+ Diffuse membranous
− May be focally positive in high-grade tumors
− May be focally positive in high-grade areas
−
−
−
−
−
+ Nuclear
+ Membranous
Multilocular cystic RCC
+
+
−
+ Diffuse membranous
+ May be patchy
−
−
−
−
−
−
+ Nuclear
+ Membranous
Clear cell papillary RCC
+
+
−
+ Membranous sparing the luminal border
+ Diffuse
−
−
−
−
−
−
+ Nuclear
− May be positive focally
Chromophobe RCC
+
+
− Rarely focally positive
− May be positive adjacent to necrosis
+
−
+
−
−
−
−
+ Nuclear
− May be focally positive in granular cells
Oncocytoma
+
+
−
−
−
− May be focally positive
+
−
−
−
−
+
− May be focally positive
Xp11 translocation-assorted carcinoma
−
− May be focallypositive
−
− May be focally positive
−
−
−
+ Nuclear
−
− Rarely focal positivity
− Rarely focal positivity
− May be patchy positive
+ Membranous and cytoplasmic
6;11 translocation-associated carcinoma
−
− May be focally positive
−
− May be focally positive
−
−
−
−
+ Nuclear
+ In 50% of cases usually patchy
+ In 50% of cases usually patchy
− May be patchy positive
+/− Membranous and cytoplasmic
Angiomyolipoma
−
−
+ May be negative in the epithelioid areas
− May be positive adjacent to necrosis
−
−
−
−
−
+ Focal or diffuse
+ Focal or diffuse
−
−
FIGURE 39A.1. Clear cell renal cell carcinoma. (A) Notice the rich vascular network lining nests and acini. The clear cell cytology, growth pattern, and prominent vascularity may be altered in high-grade lesions. (B) Diffuse membranous immunoreactivity for CA9. (See color insert.)
Many investigators have attempted to establish grading schemes for RCC with mixed results. In 1981, Fuhrman and her collaborators published a four-tiered classification and showed good correlation with clinical outcome in clear cell tumors (11). Grade I tumors have small (<7 μm), regular hyperchromatic nuclei without nucleoli. At the other extreme, grade 4 tumors had markedly enlarged pleomorphic nuclei with prominent and irregular nucleoli. Despite the fact that this classification is very cumbersome to apply in general practice (due in great part to the fact that it depends on the actual measurement of neoplastic nuclei), we have found it to be very useful in predicting clinical behavior, but only in cases of clear cell carcinoma. Tumors with sarcomatous features, irrespective of its tumor type of origin, should be considered high grade. Other features that correlate with outcome include size, pathologic and clinical stage at presentation, as well as microscopic necrosis (36,37,38).
FIGURE 39A.2. Clear cell renal cell carcinoma. The cells with high-grade nuclei often lack clear cytoplasm, which in turn becomes eosinophilic or basophilic. (See color insert.)
Clear cell renal cell carcinomas exhibit diffuse membranous immunoreactivity for CAIX as well as CD10. They are negative for CK7, racemase, and CD117 (Table 39A.3). Clear cell RCCs are characterized by the loss of genetic material of the short arm of chromosome 3 (3p) and mutations in VHL gene (39,40,41,42,43). In patients with von Hippel-Lindau disease, such losses and mutations are described in virtually all cases. Interestingly, somatic mutations/hypermethylations in the same region can be found in 75% to 80% of the more common sporadic, unilateral and unifocal tumors as well (44).
Multilocular Cystic Renal Cell Carcinoma
Multilocular cystic renal cell carcinoma represents a rare variant of clear cell (conventional) renal carcinoma. They constitute between 3.1% and 6% of clear cell RCC (45,46,47). They have been described in adult patients with a mean age of 51 years at diagnosis. Although the available follow-up has been somewhat limited, none of the reported cases have progressed. Recently, it has been shown that these tumors are associated with silencing of the VHL gene similar to conventional clear cell carcinomas. The gross appearance is that of a well-circumscribed, multicystic mass that is separated from the adjacent renal parenchyma by a fibrous pseudocapsule. The cysts are lined by thin fibrous septa, are variable in size, and may contain serous or bloody fluid or clot. No solid or expansile masses of tumor are present.
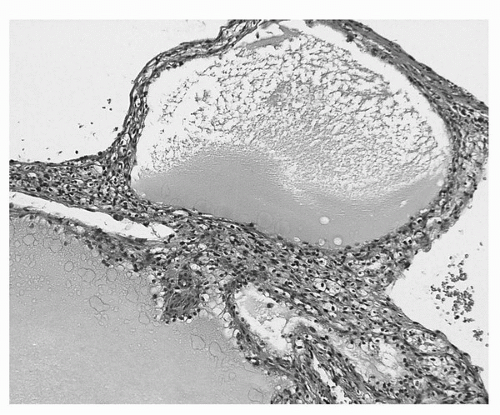
Microscopically, the thin fibrous septa are lined by one or several layers of neoplastic epithelial cells (Fig. 39A.3). One must be aware that conventional clear cell renal cell carcinomas may be partially cystic and contain thin fibrous septa virtually identical to what is seen in multilocular cystic renal cell carcinoma. The key to the correct diagnosis is the presence of expansile masses of clear cells either adjacent or within the cystic component. There is no agreement as to the size of the expansile mass required to classify the tumor as a conventional clear cell carcinoma. However, in order to maintain the correlation to benign clinical outcome, we do not accept any expansile mass in tumors classified as multilocular cystic renal cell carcinoma, irrespective of its size.
FIGURE 39A.3. Multilocular cystic renal cell carcinoma. Multiple thin-walled cysts without expansile masses of tumor. The cysts are lined, at least focally, by a single layer of cells with clear cytoplasm, while the thin septa contain nests and clusters of clear cells. (See color insert.)
CLEAR CELL PAPILLARY RENAL CELL CARCINOMA
“Clear cell papillary RCC” was recently described by Tickoo et al. in the setting of end-stage kidneys, although we now know that it may occur without evidence of impaired renal function (48,49,50). Originally believed to be a morphologic variant of conventional clear cell carcinoma, published series as well as our experience reveals no evidence of 3p25.3 losses or VHL gene mutations, making it a distinct entity. Additionally, polysomy of chromosome 7 is not seen, as would be expected in many papillary renal cell carcinomas. These tumors are usually unicentric, unilateral and small, the largest described being 5 cm. To date, no case has been documented to metastasize, although the number of cases reported and the amount of follow-up information is limited. It is common for the lesion to be cystic or partially cystic and partially solid. Microscopically, the tumor cells have uniformly clear cytoplasm and low-grade nuclei. The cells are cuboidal and arranged in tight tubular/acinar structures, particularly in the tumor-bearing solid areas (Fig. 39A.4). Invariably clear cells also line small, tightly packed papillary structures. Characteristically, the tumor nuclei are arranged in a linear fashion, away from the basal aspect of the cell, either in the center of the cell or apically. They have a unique pattern of antigen expression. Like clear cell carcinoma, they have diffuse membranous expression of CAIX which occasionally spares the luminal border of the cell (cup-like). They also express CK7 but not racemase. CD10 is either negative or only focally positive (Table 39A.3).
Papillary Renal Cell Carcinoma
Papillary renal cell carcinoma is an enigmatic group, which includes mostly the cases previously considered to be tubulopapillary type (51). We now know that there are at least two and certainly several more distinct types of papillary RCC, both at the morphologic and genetic level. Some appear to have distinct clinical behaviors as well. As a group they constitute approximately 13% of all renal cortical neoplasms. During the last decade, pathologists have classified papillary tumors into type 1 and type 2, based on their cytologic and architectural features. The “classic” or type 1 tumors have scant or moderate amounts of amphophilic or basophilic cytoplasm and low-grade nuclei (Fig. 39A.5). Type 2 tumors have more abundant eosinophilic cytoplasm, larger, high-grade, and mutilayered nuclei with prominent nucleoli (Fig. 39A.6). As you might expect, most of these tumors have a papillary, tubular or tubulo-papillary growth pattern. Some tumors have a “solid” growth pattern due to compression of the papillary structures, making them difficult to identify. Other tumors have a striking “glomeruloid” appearance. The most common type is likely to be multifocal and bilateral, with one or several dominant masses and other minute tumors throughout the cortex (socalled papillary-tubular adenomas). Papillary tumors, particularly type 1, often exhibit abundant lipid laden macrophages within the interstitium of the fibrovascular cores. This feature is very helpful in making the correct diagnosis. One must be very careful since some clear cell RCC may undergo focal necrosis in which the viable cells are seen adjacent to a blood vessel, thus producing a pseudopapillary appearance. Since this phenomenon is likely to be seen in higher-grade tumors, it is very likely that the tumor cells do not exhibit a clear cytoplasm, confounding the problem. Again, looking at many areas of the lesion will allow us to classify these tumors correctly.
Only gold members can continue reading. Log In or Register to continue