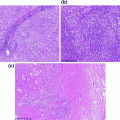
Fig. 30.1
Normal bone marrow. Normal elements of the bone marrow include megakaryocytes, erythroid cells and myeloid cells. Megakaryocytes are large cells with multilobated nuclei. Erythroid cells show dark round nuclei that condense and are extruded as the cells mature. Myeloid cells show indented and segmented nuclei, with variable granularity. Hematoxylin and eosin stain, original magnification ×400 (a) and ×1000 (b)
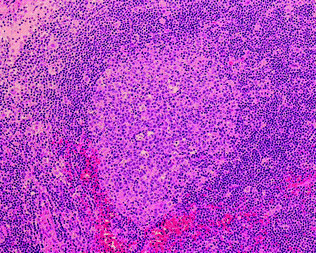
Fig. 30.2
Normal lymph node. This section shows the quintessential lymph node structure, a follicle with a germinal center. The germinal center is the central region of a follicle, with a mixture of small and large cells, mitotic figures and tingible body macrophages. Surrounding the germinal center is a mantle zone, a compact region of small lymphocytes. Between follicles are interfollicular zones, which can be appreciated at the edges of the photomicrograph. Hematoxylin and eosin stain, original magnification ×400
30.1.2 Lymphoid Activation and the Basis of Pathogenetic Translocations
To understand the cell of origin in lymphoma, it is necessary to consider lymphoid activation. B-cells begin their lives in the bone marrow as precursors, and as they mature they move out into the periphery where they populate the lymph node cortex. When antigenic stimulation leads to activation of the immune system, the follicles in the lymph nodes develop germinal centers. In these germinal centers, B-cells undergo somatic hypermutation and class-switch recombination, and some eventually develop into plasma cells capable of producing specific immunoglobulins. Both processes constitute tightly controlled DNA mutation of the immunoglobulin loci, catalyzed by specific enzymes including activation-induced deaminase (AID) [3]. During, this maturation process and controlled DNA mutation, most of the B-cells acquire mutations that do not increase their affinity for antigen, and undergo programmed cell death. A subset survives which is selected for an increased affinity of their immunoglobulin for specific antigens. Occasionally, however, these mechanisms of control and selection fail, and a B-cell with a pathological DNA mutation survives the germinal center reaction with a potential for additional genetic events and neoplastic transformation. Based on the stage of differentiation of the cell that acquires the mutation, different subtypes of lymphoma can develop (Fig. 30.3).
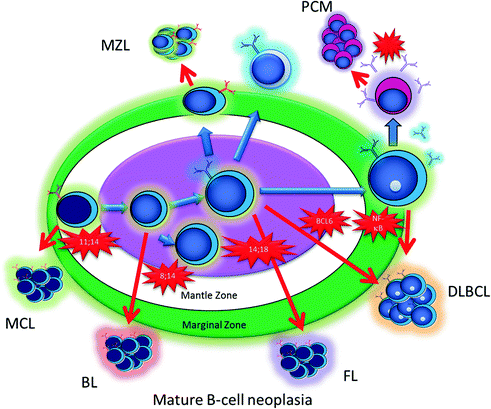
Fig. 30.3
A schematic of antigen-driven B-cell maturation and pathologic translocations that can lead to the different subtypes of B-cell lymphoma. Abbreviations: MCL, Mantle cell lymphoma; BL, Burkitt Lymphoma; FL, Follicular lymphoma; DLBCL, Diffuse large B-cell lymphoma; PCM, Plasma cell myeloma; MZL, Marginal Zone lymphoma
Many genetic alterations involve breaks in the immunoglobulin heavy chain (IGH) gene locus on chromosome 14 which can result in a translocation with an oncogene such as MYC [4]. This results in a balanced translocation and abnormal expression of MYC protein. MYC is a powerful oncogene which drives cells into the cell cycle. In cells with t(14;18), MYC is juxtaposed between the IGH locus, which is highly active in B-cells, resulting in a massive overexpression of this oncogene which in turn drives cell growth and proliferation. Similarly, translocations involving the IGH gene and BCL2 are typical of follicular lymphoma (FL) [5, 6]. In this case, the IGH locus is juxtaposed against BCL2 whose overexpression prevents cell death of lymphoma cells. Abnormalities in BCL6 can arise from point mutations, and can involve the same IGH locus. BCL6 is a transcription factor and when over expressed can block B-cell differentiation resulting in long-lived proliferative cells [7]. This brief discussion illustrates neoplastic progression from increased survival or a block in apoptosis with BCL2 translocations, increased growth from oncogenes that drive cells into cell cycle like MYC, and a potential block in differentiation from genes such as BCL6 which govern the germinal center cell reaction. These three concepts, survival, growth, and differentiation, are thought to involve all lymphoid tumors to various degrees.
30.1.3 Epstein-Barr Virus and Lymphomagenesis
The infectious agent most often associated with lymphomas is the Epstein-Barr virus (EBV). Over 90 % of the population is infected with EBV by the time they reach adulthood [8], and EBV persists for the lifetime of the host in the latent form by embedding itself in a very small percentage of B-cells, where it is thought to be held in check by T-cell-mediated immunity. When there is impairment of the immune system for example due to iatrogenic drugs, transplantation, or infections such as human immunodeficiency virus (HIV), the EBV-infected B-cells proliferate and are subject to additional genetic alterations which can lead to lymphoma. In an EBV-positive lymphoma not only is the B-cell clonal, but the EBV within that B-cell is clonal as well. In patients over the age of 60, lymphomatous proliferations known as EBV-positive diffuse large B-cell lymphoma (DLBCL) of the elderly may occur, presumably related to senescence of the immune system [9]. Immune dysregulation is clearly a key factor, which contributes to lymphomagenesis in many circumstances. Immune senescence, geography, congenital, or acquired immune defects may all contribute to the neoplastic process.
30.2 Neoplastic Lymphoid Proliferations
Given the breadth of hematologic malignancy subtypes, this discussion will be restricted to mature lymphoid neoplasms, which tend to involve the lymph nodes and other tissues, but the malignant cells may also circulate in the peripheral blood as leukemias. These neoplasms can also be classified as Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL), the latter a large group derived from mature B-cells, T-cells, or NK cells. B-cell NHL is far more common than T-cell NHL, and comprises approximately 80 % of lymphomas in Europe and the United States [10]. Although heterogeneous in their etiology and pathogenesis, many are derived from the germinal center reaction as mentioned above, and certain types are associated with infectious agents. In the following discussion, we will briefly introduce HL and T-cell NHL, and summarize the relevant immunological considerations and molecular pathogenesis of the most common B-cell NHL subtypes (also summarized in Tables 30.1 and 30.2) [11–22].
Table 30.1
A partial list of b-cell lymphoma with immunohistochemical staining patterns
Lymphoma type | CD20 | CD5 | CD10 | CD23 | BCL1 | BCL2 | BCL6 |
|---|---|---|---|---|---|---|---|
Mantle cell lymphoma | + | + | − | − | + | − | − |
CLL/SLL | + | + | − | + | − | + | − |
Marginal zone lymphoma | + | − | − | − | − | + | − |
Burkitt lymphoma | + | − | + | − | − | − | + |
Follicular lymphoma | + | − | + | ± | − | + | + |
DLBCL, GC type | + | − | + | − | − | ± | ± |
DLBCL, non GC type | + | − | − | − | – | ± | ± |
Table 30.2
Molecular genetic abnormalities seen in lymphoid malignancies
Lymphoma | Molecular alterations |
|---|---|
Small lymphocytic lymphoma/CLL | Del 13q14.3 most common followed by trisomy 12 and deletions of 11q22–23, 17p13 and 6q21 [11]. Possible targets in the 13q14.3 region are two microRNA genes, miR-16–1 and miR-15a, ATM in the 11q22–23 region and TP53 in the 17p13 region |
Follicular lymphoma | Translocation t(14;18)(q32;q21) and BCL2 gene rearrangements are the hallmark of follicular lymphoma [12]. Abnormalities of 3q27 and/or BCL6 rearrangement are found in 5–15 % of cases Other genetic alterations include loss of 1p, 6q, 10q and 17p, and gains of chromosomes 1, 6p, 7, 8, 12q, X among others |
Plasma cell myeloma | Multiple numerical and structural abnormalities are found, including trisomys, whole or partial chromosome deletions and translocations [13]. Complex cytogenetic abnormalities are common. The most frequent chromosome translocations involve the heavy chain locus (IGH@) on chromosome 14q32. Major recurrent oncogenes involved in 14q32 translocations include cyclin D1 (11q13) |
Mantle cell lymphoma | The t(11;14)(q13;q32) between IGH and cyclin D1 (CCND1) is the hallmark of mantle cell lymphoma [14], and is found in about 95 % of cases. MCL also carries a high number of non-random secondary chromosomal abnormalities including loss of TP53 and trisomy 12 |
Diffuse large B-cell lymphoma | Most commonly there are abnormalities, including translocations, involving the 3q27 and the BCL6 gene [15]. Rearrangements of the MYC gene occur in about 10 %, and are usually associated with a complex pattern of additional molecular alterations. Other mutations in DLBCL include frequent mutations in MYD88, CD79A/B, CARD11, A20 loss, and TP5 3 [16] |
Burkitt lymphoma | The molecular hallmark of BL is a translocation involving the MYC gene at band q24, from chromosome 8 to the Ig heavy chain region on chromosome 14 [t(8;14)] or less commonly at light chain loci on 2p12 [t(2;8)] or 22q11[t(8;22)] [17] |
Marginal zone B-cell lymphoma (MZL) | Approximately 30 % of splenic MZL show 7q deletion Splenic marginal zone lymphomas may have mutations involving NOTCH2. Chromosomal translocations associated with extranodal marginal zone lymphomas (lymphomas of mucosal associated lymphoid tissues or MALT) include t(11;18), t(1;14), t(14;18) and t(3;14), resulting in the production of a chimeric protein (API2-MALT1) [18]. Trisomy’s including chromosome 3 and 18 also occur |
Hairy cell leukemia (HCL) | BRAFV600E mutations are the most important genetic abnormalities which characterize HCL [19] |
Peripheral T-cell lymphoma (PTCL) | PTCL tend to have multiple abnormalities and complex karyotypes [20]. Recurrent chromosomal gains have been observed in chromosomes 7q, 8q, 17q and 22q, and recurrent losses in chromosomes 4q, 5q,and 12q among others. PTCL involving T-follicular helper cells, such as angioimmunoblastic T-cell lymphoma (AITL), have frequent mutations involving RHOA, TET2, and DNMT3A |
Anaplastic large cell lymphoma (ALCL) | The hallmark of ALK positive ALCL is fusion of the ALK gene with various translocation partners. The most frequent genetic alteration is a translocation, t(2;5)(p23;q35), between the ALK gene on chromosome 2 and the nucleophosmin (NPM) gene on chromosome 5 [21]. Variant translocations involving ALK and other partner genes occur less frequently. In ALK negative ALCL the DUSP 22 translocation characterizes an important subgroup with improved outcome, while those with TP63 do poorly [22] |
30.2.1 Hodgkin Lymphoma and T-Cell Lymphoma
Together, HL and T-cell NHL constitute a minority of cases of lymphoma in the United States and Europe, but their incidence differs in other parts of the world [10]. Briefly, HL was the first defined type of lymphoma in the early nineteenth century, and it was recognized as such due to a unique and stereotyped presentation and anatomic pattern of involvement. Many years later, histopathologists identified the malignant cell, the “Reed-Sternberg” (RS) cell that has a characteristic morphologic appearance (Fig. 30.4). Histologically, HL contains a minority of these neoplastic cells with an accompanying infiltrate composed of a diverse group of immune cells including histiocytes, plasma cells, small lymphocytes, and eosinophils. Molecular studies of singly isolated RS cells revealed that these were cells of B-cell origin that had undergone crippling mutations of their immunoglobulin genes and activation of NFκ[kappa]B. Moreover, recent studies have identified that amplifications in 9p24 led to overexpression of the genes encoding the immunoregulatory proteins PD-L1 and PD-L2, and this is now the basis of a novel therapeutic approach [23]. HL can be divided into two major subgroups including nodular lymphocyte predominant HL, where tumor cells retain immunophenotypic features of germinal center B-cells, and classical HL, which fail to express these gene products. Classical HL is further subdivided into nodular sclerosis classical HL, mixed cellularity classical HL, lymphocyte-rich classical HL and lymphocyte depleted classical HL, definitions of which are beyond the scope of this book chapter. The reader can refer to a recent review of this disease for further information [24].
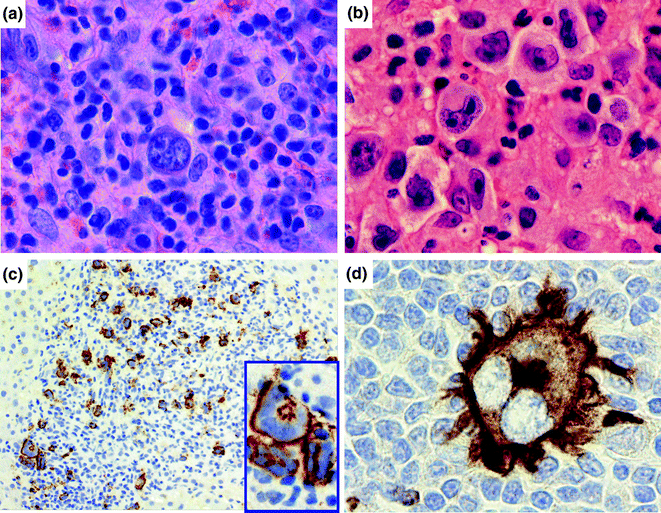
Fig. 30.4
Classical Hodgkin lymphoma. a, b Histologically, classical Hodgkin lymphoma is composed of a mixture of cells, including small lymphocytes, plasma cells, histiocytes, and eosinophils, as well as the large, multilobated Reed-Sternberg (RS) cell. Hematoxylin counterstain, original magnification ×1000. c The RS cell classically stains for the activation marker CD30 (immunohistochemistry with anti-CD30 antibody, hematoxylin counterstain, original magnification, ×200 and inset, ×1000). d The RS cell also stains for CD15, a myeloid related antigen (immunohistochemistry with anti-CD15 antibody, hematoxylin counterstain, original magnification, ×1000)
Another major lymphoma subtype is T-cell NHL. T-cell lymphomas constitute a diverse array of lymphoma types that encompass different cells of origin, many different mutations, and show a range of histologic features from very bland to highly pleomorphic. Recent progress in the field has focused on more specific definitions of subtypes of T-cell lymphoma that may have a specific histogenesis. For example, Angioimmunoblastic T-cell lymphoma (AITL) is thought to derive from a follicular T-helper cell, which shows expression of specific markers including PD1 and CXCL13. The proliferation of the T-cells leads to recruitment of additional immune cells and vascular proliferation, generating the classic appearance of this disease (Fig. 30.5). In addition, there are scattered B-immunoblasts within the infiltrate that are generally positive for EBV, and the latter may represent the underlying etiological agent responsible for this disease. Other forms of T-cell lymphoma include anaplastic large cell lymphoma and peripheral T-cell lymphoma, along with a number of others. In general, T-cell lymphoma is thought to be more common in Asian populations, where an association with EBV has been noted [10].
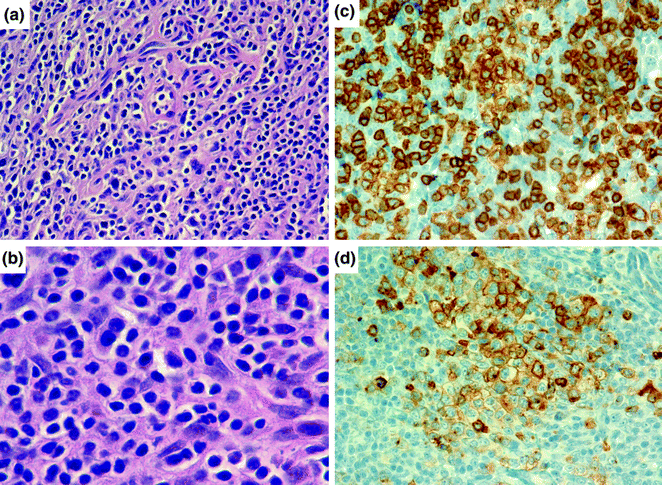
Fig. 30.5
Angioimmunoblastic T-cell lymphoma. a, b AITL consists of a mixture of cells, including malignant T-cells and a proliferation of high endothelial venules and follicular dendritic cells. The T cells generally show “water-clear cytoplasm” as can be appreciated in b. Hematoxylin and eosin stain, original magnification, ×200 (a) and ×1000 (b). The T cells stain with antibodies to CD3e (c) and to CD10 (d), a follicular marker, pointing to a T-follicular helper cell origin for these lymphomas. Hematoxylin counterstain, original magnification ×400
30.2.2 Non-Hodgkin B-Cell Lymphoma
30.2.2.1 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) is a neoplastic proliferation comprised of small regular lymphoid cells similar to those found in normal tissues but present in increased numbers and effacing normal architecture in the bone marrow, peripheral blood, lymph nodes, spleen, and other organs (Fig. 30.6). CLL is defined as greater than 5 × 109 L circulating clonal B-cells with appropriate morphology and immunophenotype. When the disease presents in lymph nodes it is called SLL. CLL has a relatively high genetic predisposition, with a family history of CLL in up to 10 % of cases. Immunoglobulin genes are rearranged and with somatic hypermutation in 50–60 % of cases [25], the remainder has unmutated immunoglobulin genes. Most cases have cytogenetic abnormalities, particularly del (13q) in about 50 % of cases, trisomy 12, as well as deletions of 11q, 17p13, and 6q21. For many years the candidate tumor suppressor on chromosome 13q was unknown, as the deleted region did not show many protein coding genes. However, the recognition of non-coding RNA and microRNA in the last decade led to the identification of a candidate tumor suppressor microRNA gene on chromosome 13q [26]. Recent work in mouse models has suggested that this microRNA, miR-15a/16 may be pathogenetic in causing low-grade B-lymphocytosis and eventually CLL/SLL [27]. In addition to the pathogenetic implications, many of these genetic findings have prognostic implications. CLL patients with mutated IGH loci have a better prognosis, whereas those with deletions of 17p in the region of the TP53 tumor suppressor gene generally have a worse prognosis.
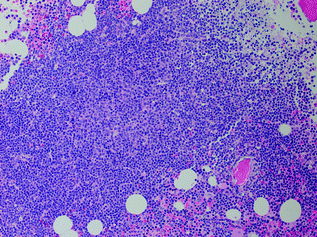
Fig. 30.6
Bone marrow involved by chronic lymphocytic leukemia. There is an aggregate of small neoplastic lymphoid cells with a central proliferation center. Hematoxylin and Eosin stain, original magnification ×400
30.2.2.2 Burkitt Lymphoma
Burkitt lymphoma (BL) is an example of lymphomagenesis with specific molecular alterations, typically but not necessarily associated with EBV. It is the most common non-Hodgkin lymphoma in children and adolescents. Pathologically, it is a monomorphic proliferation of medium sized transformed germinal center related B-cells with round nuclei, clumped chromatin, basophilic cytoplasm, squared-off cell borders, cytoplasmic vacuoles, medium-sized paracentral nucleoli, and a starry sky pattern caused by the presence of numerous phagocytic histiocytes (Fig. 30.7). Translocations involving MYC are characteristic but not specific for BL. Although greater than 90 % of African BL cases are positive for EBV, only about 30 % of cases in Europe and the United States contain EBV [28].
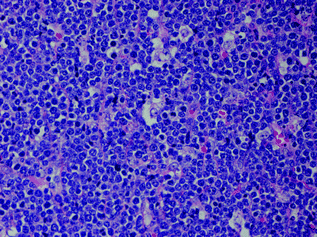
Fig. 30.7
Burkitt lymphoma showing a diffuse proliferation of cohesive intermediate sized cells with distinct nucleoli and frequent mitoses. There are scattered phagocytic histiocytes lend a starry sky appearance to the infiltrate. Hematoxylin and Eosin stain, original magnification ×400
BL has a characteristic chromosome abnormality of translocation at chromosome 8q24 involving MYC usually with chromosome 14q32 involving IGH [4, 29–31]. Variant translocations occur with the lambda light chain gene (IGL) at chromosome 22q11 or the kappa gene (IGK) at chromosome 2p12 in up to 16 % of cases [32–34]. The MYC break-apart probe is generally used in interphase fluorescence in situ hybridization (FISH) to detect the translocations, since it is not dependent on the specific translocation partner. The breakpoints may vary in different types of BL. For example, in endemic BL the MYC breakpoint is generally far 5′ centromeric of MYC, while in HIV-related BL the breakpoint tends to occur between exon and intron 1. Nonetheless, it is thought that all of these translocations result in a massive, unregulated overexpression of MYC.
MYC and Its Role in Burkitt Lymphoma
Recent studies have suggested that MYC is a global amplifier of all active promoters and enhancers in the genome, rather than a conventional transcription factor [35]. In this setting, MYC is able to drive proliferation by increasing glucose utilization and increasing protein synthesis. However, this interpretation of the data has been questioned by a second, more recent study, which reiterates the capacity of MYC to target specific genes and drive tumorigenesis [36]. Nonetheless, it is apparent that neither EBV infection nor MYC translocation are sufficient to initiate and maintain neoplastic proliferations in BL, and t(8;14) has even been detected in normal individuals [37]. There is considerable new data regarding additional genetic aberrations which contribute to the pathogenesis of BL. For example, co-activation of MYC-PI3K selects for stabilizing mutations in cyclin D3 (CCND3), which is a key regulator of the cell cycle in germinal center B-cells [38], and cyclin D3 is commonly overexpressed in BL. Abrogating PI3K signaling or cyclin D3 leads to BL cell death [39], highlighting the importance of this pathway and providing a potential for use of therapeutic agents that target the PI3K signal transduction cascade. Thus, the relationship between c-MYC and PI3K signaling has far-reaching effects in BL diagnosis, prognosis, and treatment and bears further study.
30.2.2.3 Lymphoma of Mucosal Associated Lymphoid Tissues
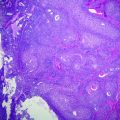
Mucosal associated lymphoid tissue (MALT) lymphomas typically involve extranodal tissues including the stomach, orbit, skin, thyroid, and other sites. It is comprised of small B-lymphocytes most commonly resembling those in the marginal zone of the lymphoid follicle, often called centrocyte-like cells. The cells are small with mildly irregular nuclei and increased often clear cytoplasm. They frequently show plasma cell differentiation. Where, they involve epithelial sites the neoplastic cells typically infiltrate the adjacent epithelium forming so-called lymphoepithelial lesions (Fig. 30.8).
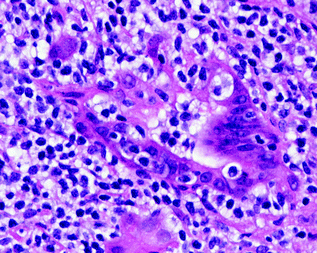
Fig. 30.8
MALT lymphoma of the stomach. The neoplastic cells are small with mildly irregular nuclei and clear cytoplasm. They are infiltrating a gastric gland forming a lymphoepithelial lesion. Hematoxylin and Eosin stain, original magnification ×400
MALT lymphomas serve as a model of lymphomagenesis related to infection, antigenic stimulation, and genetic dysregulation. The clearest example occurs in the case of gastric MALT lymphomas which are often preceded by gastritis due to infection with the bacterial organism Helicobacter pylori. This causes an inflammatory gastritis which includes formation of reactive germinal centers, and over time genetic abnormalities can result in an uncontrolled clonal proliferation of B-cells whose antigen receptors recognize H. pylori. This process can be reversed and the lymphoma eradicated by using antibiotics effective against H. pylori [40]. There are several genetic abnormalities which characterize MALT lymphomas including the translocation t(11;18), t(14;18)(q32;q21) not involving the BCL2 gene, and t(3;14) which results in the production of a chimeric protein known as API-MALT1 [41]. Many of the translocations are thought to result in the constitutive activation of the NFκ[kappa]B pathway, which results in increased cell growth and inhibition of apoptosis. In MALT lymphoma there are different genetic abnormalities associated with various sites of disease and geographic variability, and many of these abnormalities have prognostic implications including the likelihood of responding the antibiotic therapy. The latter therapy is one of the few examples where removal of an inciting agent can result in the regression of a malignancy, and highlights the close relationship between normal immune system function and development of B-cell neoplasia.
30.2.2.4 Diffuse Large B-Cell Lymphoma
DLBCL is a common histologic subtype of lymphoma and comprises 30 % of adult non-Hodgkin lymphomas in the west and an even higher percentage in developing countries [10]. DLBCL is an aggressive form of lymphoma and despite improved treatment regimens about 40 % of patients fail to respond or relapse following chemotherapy. DLBCL is generally characterized by clonal rearrangements of IGH, IGK, and IGL genes, with many cases also showing somatic hypermutation, indicative of a post-germinal center cell origin. The most common cytogenetic findings are abnormalities in chromosome 3q27 involving BCL6 in 30 % of cases, abnormalities of BCL2 with translocation t(14;18) in 20 %, and MYC rearrangement in 10 % of cases [42, 43]. DLBCL can occur at nodal or extranodal sites. There is an ongoing effort to tailor therapy to individual patients with subtypes of DLBCL, and prognostic markers are becoming increasingly important.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


