Fig. 22.1
Anatomy of the colon and rectum with segments labeled. Peritonealized and non-peritonealized segments are shaded different colors
The transverse colon is suspended from a sheet of peritonealized fat, the lesser omentum, which originates along the greater curvature of the stomach. Hanging from the surface of the transverse colon is a second larger sheet of peritonealized fat, the greater omentum. When staging and assessing the adequacy of resection of CRC (particularly the status of the radial resection margin), it is important to know which anatomic segments are involved by tumor and whether a peritoneal lining covers these segments. The cecum is entirely peritonealized, while the ascending colon, hepatic and splenic flexures, and descending colon are only covered by peritoneum along their anterolateral surfaces. The transverse and sigmoid colon are almost completely surrounded by peritoneum, except at their mesenteric attachments. The upper two-thirds of the rectum is covered laterally and/or anteriorly by peritoneum, whereas the lower third is below the peritoneal reflection and therefore has no peritoneal covering.
The blood supply to the cecum, ascending, and proximal transverse colon comes from branches of the superior mesenteric artery, while branches of the inferior mesenteric artery supply the remainder of the colon and the proximal rectum (Fig. 22.1). The distal rectum is supplied by the internal iliac and pudendal arteries [8]. Venous drainage of the colon and rectum is to the portal vein via the superior and inferior mesenteric veins. Lymph nodes are located within the pericolorectal connective tissue, as well as along the branches of the mesenteric vessels. From here, lymphatic drainage converges on major nodal groups around the roots of the superior and inferior mesenteric arteries, ultimately emptying into the thoracic duct. The lymphatic drainage of the distal rectum is to the hypogastric, obturator, and internal iliac nodes via the paraaortic nodes [8].
22.4 Histology of the Benign Colon and Rectum
Figure 22.2 illustrates the layers of the colorectum, which, from inner (luminal) to outer surface, are the (1) mucosa, composed of the epithelium, lamina propria, and muscularis mucosae, (2) submucosa, (3) muscularis propria, with inner circular and outer longitudinal muscle layers, (4) pericolorectal adipose tissue or subserosa (subserosa being present in the peritonealized segments of colon and rectum, and (5) serosa, the outermost peritonealized wrapping of the intestine (where present). The luminal surface of the large intestine is lined by straight, nonbranching crypts oriented parallel to one another and perpendicular to the lumen, like test tubes in a rack. The epithelial cells lining these crypts are of two main types (Fig. 22.3a): (1) absorptive enterocytes, which are tall, columnar cells with basally oriented, ovoid to elongated nuclei, pale eosinophilic cytoplasmic mucin, and an apical microvillus brush border, and (2) mucus secreting goblet cells, so named because they resemble drinking goblets in shape, which are interspersed between the absorptive enterocytes and contain small, basally oriented nuclei and cytoplasm distended by pale basophilic acid mucin, which stains positive by Alcian blue at pH 2.5 (in contrast to the neutral mucin present in absorptive enterocytes, which is negative with Alcian blue and stains magenta on a periodic acid Schiff stain). The mucus secreted by goblet cells forms a protective layer against luminal pathogens.
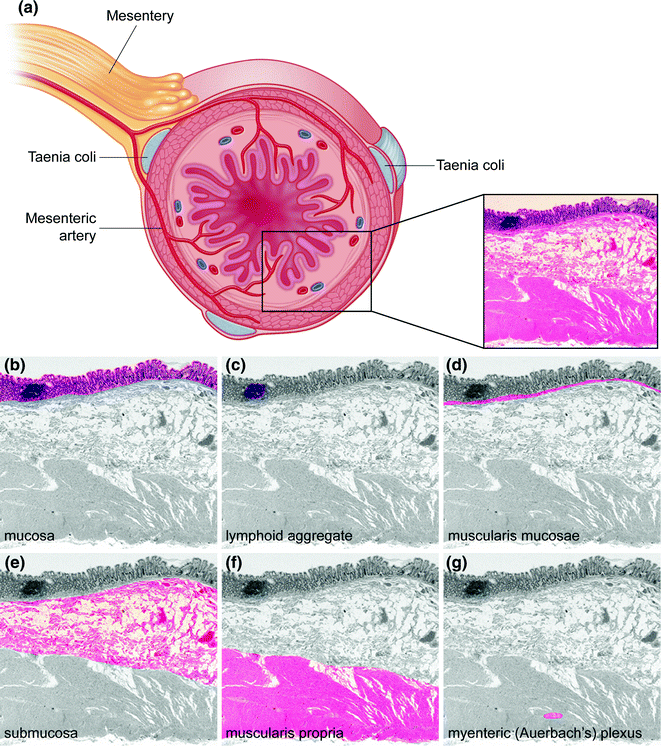
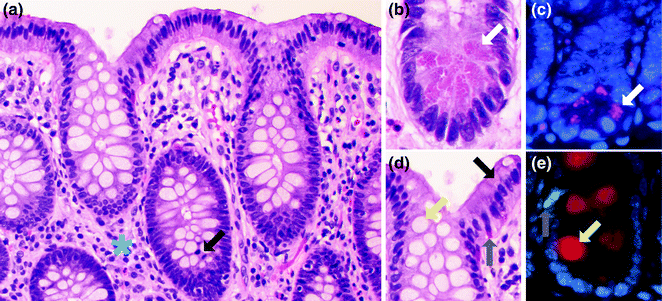
Fig. 22.2
Histology of the colon. a Cross section of intestinal wall illustrating the different histologic layers. Tissue is stained with hematoxylin and eosin (H&E). b–g Histologic layers of the colon. In each panel, a single histologic portion is highlighted in color
Fig. 22.3
Cell types of the normal colorectum. a H&E stain of normal colon. A mitotic figure is highlighted by a black arrow. The blue asterisk is in the lamina propria. b High magnification view of the bottom of the colonic crypt. A Paneth cell is highlighted with a white arrow. c Immunofluorescence for lysozyme (purple), a marker of Paneth cells. The Paneth cell is highlighted with a white arrow. d High magnification view of the top of the colonic crypt. A goblet cell is highlighted with a yellow arrow. Enterocytes (black arrow) and enteroendocrine cells (gray arrow) are also highlighted. e Immunofluorescence for mucin 2 (red), a marker of goblet cells and serotonin (green), a marker for some enteroendocrine cells. The goblet cell is highlighted with a yellow arrow. The enteroendocrine cell is highlighted with a gray arrow
Another cell type normally present within the epithelium, and found at the crypt base, is the endocrine cell, of which there are two morphologic types: (1) the enterochromaffin cell (Fig. 22.3b), which secretes serotonin and is easily identified on hematoxylin and eosin (H&E) stained sections as having a pyramidal or spindled shape, a luminally oriented nucleus, and basally oriented, reddish-orange granules, and (2) the non-enterochromaffin endocrine cell, which is not easily identified on H&E stained sections, and has a basally located nucleus and inconspicuous, apically oriented granules. Endocrine cells secrete various peptides that include somatostatin, cholecystokinin, motilin, secretin, vasoactive intestinal polypeptide (VIP), substance P, glucagon, neurotensin, and gastric inhibitory polypeptide, among others, with differences in the distributions of these cell types depending on location in the colon and rectum [8].
Also present within the bases of the crypts are Paneth cells (Fig. 22.3c), characterized by luminally oriented eosinophilic granules that contain antimicrobial proteins (defensins). Paneth cells play an important role in the innate and adaptive immune responses against luminal bacteria, and express two main types of innate pattern recognition receptors: the transmembrane toll-like receptors (TLRs) and the cytosolic nucleotide-binding oligomerization domain-2 (NOD2) receptors, both of which recognize highly conserved pathogen-associated molecular patterns (PAMPs) [10]. Paneth cells are normally found in the right colon, but may be seen in the left colon in the setting of chronic inflammation and injury. In humans, defects in Paneth cells have been linked to diseases such as neonatal necrotizing enterocolitis and inflammatory bowel disease [11, 12].
The last differentiated cell type characteristically lining the colorectum is the microfold (or M) cell, which is often found above mucosal lymphoid aggregates and functions to transport luminal antigens into the underlying lymphoid aggregate for recognition by the immune system [13, 14].
Colonic stem cells, morphologically distinctive cells in the crypt base (often called crypt base columnar cells or CBCs), can be identified by expression of LGR5/GPR49, a G protein-coupled receptor that binds the Wnt agonist R-spondin and potentiates Wnt signaling [15–18]. Each stem cell divides to yield a daughter stem cell and a committed progenitor cell, sometimes referred to as a transit amplifying cell (TAC) due to its rapid proliferation rate. These TACs can be identified by their incorporation of bromodeoxyuridine (BrdU) or tritiated thymidine, or by Ki-67 immunostaining. They are located along the sides of the crypts, immediately above the Paneth cells. As these cells proliferate and mature, they migrate upward along the crypt wall, eventually reaching the surface, where they differentiate into absorptive enterocytes, goblet cells, endocrine cells, and M cells. Some of these cells also migrate downward toward the crypt base to become Paneth cells.
Each colorectal crypt is surrounded by a basement membrane, to which the epithelial cells are anchored. The lamina propria is a supportive connective tissue stroma in between the individual crypts, which contains capillaries, small nerves, lymphatics (mainly within the deepest part of the lamina propria), and an inflammatory infiltrate containing plasma cells, histiocytes, lymphocytes, eosinophils, mast cells, and the rare neutrophil. The muscularis mucosae forms the deepest extent of the mucosa. Mucosal lymphoid aggregates are normally present within the lamina propria, sometimes extending through the muscularis mucosae and into the submucosa.
The submucosa contains loose connective tissue, blood vessels, lymphatics, nerves, and the ganglion cells of the submucosal (Meissner’s and Henle’s) plexuses. Beneath this is the muscularis propria, which contains an inner circular layer and outer longitudinal layer, between which is located the myenteric (Auerbach’s) plexus. The taenia coli are actually localized thickenings of this outer longitudinal muscle layer. External to the muscularis propria is pericolorectal connective tissue containing blood vessels, nerves, and a variable amount of fat. In segments of the colorectum that are covered by peritoneum, this layer is referred to as the subserosa, and the outermost layer, the serosa, is characterized by a single layer of mesothelial cells that forms the smooth outer surface of the intestine.
22.5 Premalignant Lesions of the Colon and Rectum
The earliest potential precursor to colorectal neoplasia is the aberrant crypt focus (ACF), a discrete focus of one or more crypts distinguishable from the surrounding normal crypts by a larger than normal crypt diameter and increased numbers of lining epithelial cells [19, 20]. ACF are invisible to the unaided eye, but may be seen under a dissecting microscope after methylene blue staining (Fig. 22.4), or by chromoendoscopy based on their unique pit patterns [21]. Multiple types of ACF exist, including non-dysplastic hyperplastic ACF and dysplastic ACF, although it is controversial whether all ACF progress to adenoma [22–24].
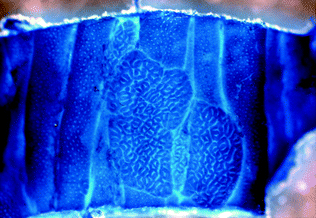
Fig. 22.4
Wholemount methylene blue staining of an aberrant crypt focus from a mouse. From Velho and Haigis [25]
The universally accepted precursor to carcinoma is dysplasia, which is formally defined as unequivocally neoplastic epithelium that is confined to the basement membrane [26]. This is in contrast to carcinoma, which represents spread of neoplastic cells beyond the basement membrane into the surrounding lamina propria (intramucosal adenocarcinoma) or beyond. Dysplasia may be grossly or endoscopically visible, or only seen on microscopic evaluation. Visible dysplasia may take on many different appearances, from polypoid lesions to ill-defined carpet-like patches, and is classified according to the Paris classification [27]. Microscopically, dysplasia is classified into several morphologic subtypes, including conventional (intestinal type), serrated, and villous/hypermucinous or gastric type [26]. Dysplasia may arise sporadically, or as a consequence of chronic inflammation, such as that seen in inflammatory bowel disease (IBD). The standardized classification used for microscopic evaluation of dysplasia (both sporadic and IBD-associated) in the United States is the Dysplasia Morphology Study Group [28], whereas the Vienna classification is used in Asia and most of Europe [29].
22.5.1 Adenomas
The most common type of dysplastic lesion is the adenoma, which often takes the gross appearance of a sessile or pedunculated polyp. However, non-polypoid flat or depressed adenomas can also occur. An estimated 12 % of individuals undergoing screening colonoscopy in the United States will develop adenomas by age 50, with the prevalence increasing up to 50 % beyond age 50 [30]. Adenomas are the classical precursor in the chromosomal instability (CIN) pathway of colorectal carcinogenesis described by Vogelstein and Fearon (see section on Molecular Pathology for details), and it is well established that endoscopic removal of these lesions reduces the risk of subsequent colorectal carcinoma [31, 32].
The morphologic subtype of dysplasia seen in adenomas is often referred to as “conventional” or intestinal type dysplasia, which may be low or high grade (Fig. 22.5). In low-grade dysplasia, the tubular architecture of the crypts is generally maintained, but the epithelial cells have enlarged, pencillate (pencil-shaped), hyperchromatic nuclei arranged in a crowded or stratified pattern. Mitotic figures and apoptotic bodies may be present. High-grade dysplasia is characterized by the presence of architectural changes such as glandular crowding and cribriform or back-to-back glands. The epithelial cells in high-grade dysplasia also appear more atypical, with more nuclear enlargement, pleomorphism, prominent nucleoli, atypical mitotic figures, and loss of nuclear polarity. Dysplasia usually extends from the crypt base all the way to the surface, but may be limited to the crypt base in early cases (“crypt dysplasia”). Although the classical adenoma-carcinoma sequence is thought of as progressing sequentially from low-grade to high-grade dysplasia and then to carcinoma, some carcinomas have been observed to arise directly from low-grade dysplasia without passing through an intermediate high-grade dysplastic stage [33].
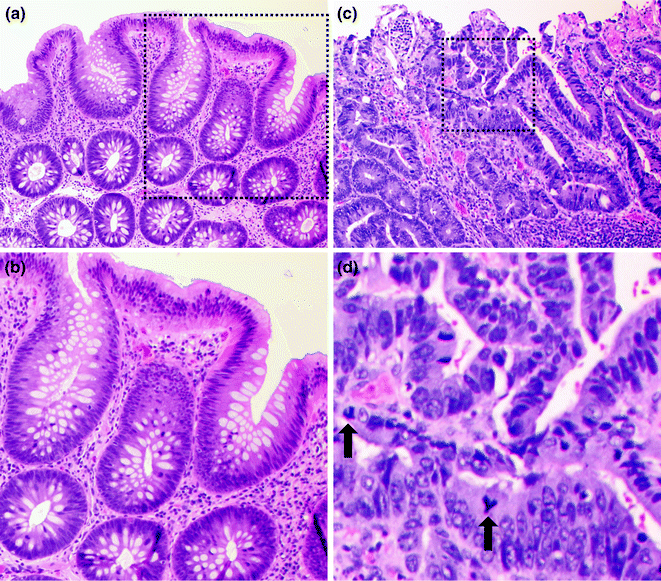
Fig. 22.5
Colonic adenoma. a Low magnification of a low grade dysplasia. b High magnification of the region outlined in black in panel A. c Low magnification of a high grade dysplasia. d High magnification of the region outlined in black in panel C
In addition to grade, adenomas are also frequently subclassified into three types based on the amount of the adenoma that exhibits a villous architecture (villi = finger or leaf-like projections). Tubular adenomas (the most common type) are 0–25 % villous, tubulovillous adenomas are 26–74 % villous, and villous adenomas are >75 % villous. The villous component appears to increase with increasing size of the adenoma [34].
Although adenomas are relatively common, most do not progress to carcinoma. Those with an increased risk of progression are referred to as “advanced adenomas,” and are defined as those that are larger than 1 cm in size, contain a villous component of >25 % (i.e., tubulovillous or villous adenomas), and/or have high-grade dysplasia [35]. For adenomas that are >1 cm in size, the estimated 10-year risk of progression to carcinoma is 10–15 % [36]. The exact rate of progression from an adenoma to carcinoma is difficult to assess because most adenomas are removed at diagnosis, but it likely varies according to the degree of dysplasia and molecular background in which the adenoma arose, with one often-cited study demonstrating a 15-year interval from the detection of an adenoma to the development of carcinoma [37].
22.5.2 Serrated Polyps
Serrated polyps comprise a heterogeneous group of neoplastic lesions that differ with regard to their morphology, predominant location within the colon (right- versus left-sided), and molecular pathology. A common feature of serrated polyps is a serrated luminal contour, thought to result from increased cellular senescence, leading to an accumulation or “piling up” of crypt epithelial cells and a serrated (sawtooth) crypt shape [38]. According to the standardized nomenclature and classification of serrated polyps proposed by the World Health Organization (WHO) Classification of Tumours of the Digestive System [39], the three main types of serrated polyps are the: (1) hyperplastic polyp, (2) sessile serrated polyp/adenoma (SSP/A), and (3) traditional serrated adenoma (TSA).
22.5.2.1 Hyperplastic Polyps
Hyperplastic polyps are, by far, the most common type of serrated polyp, comprising at least 75 % of all serrated polyps, and are present in up to 35 % of screening colonoscopy patients aged 50 or older [40]. They are sessile, often small (<5 mm) polyps that are found throughout the colorectum, but occur most often on the left side. Three morphologic subtypes of hyperplastic polyp are recognized: microvesicular (MVHP), goblet cell rich (GCHP), and mucin poor (MPHP). The most common subtype, the MVHP (Fig. 22.6), is characterized by crypts that are narrow at the base, wide at the surface, and lined by enterocytes containing fine (microvesicular) mucin droplets, interspersed with variable numbers of goblet cells. The nuclei of the enterocytes are small and arranged in a single row, without stratification or hyperchromasia. On cross-sectional analysis, the crypts of the MVHP exhibit a characteristic stellate shape. The GCHP is much less common than the MVHP, and is characterized by mildly dilated crypts lined predominantly by goblet cells, with minimal to no luminal serration. The least common subtype, the MPHP, looks like a mucin-depleted MVHP, and may, in fact, represent a MVHP with reactive changes. Although all hyperplastic polyps are considered non-dysplastic, some are precursors to other serrated polyp types (SSP/A and TSA) with established premalignant potential (see below).
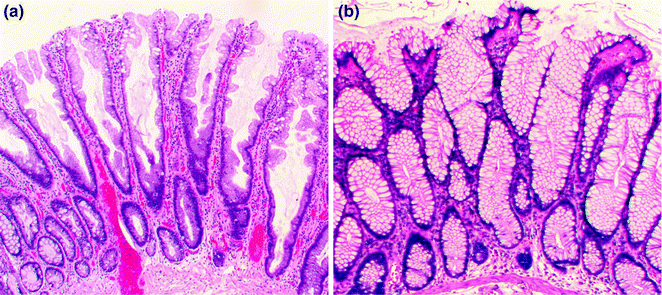
Fig. 22.6
Hyperplastic polyps. a Microvesicular. b Goblet cell rich
22.5.2.2 Sessile Serrated Polyps/Adenomas
SSP/As represent about 9–40 % of all serrated polyps and have also been referred to as “sessile serrated adenoma (SSA)” or “sessile serrated polyp (SSP)” [39, 41]. Grossly, they are sessile, usually >5 mm in size, often surfaced by a yellow mucin cap, and more commonly found in the right colon. Histologically, they resemble hyperplastic polyps, but are additionally characterized by abnormal maturation, with mature goblet cells and serration found within the crypt base, and abnormal crypt architecture, with basal dilation of crypts and a horizontal growth pattern, resulting in “boot” and “anchor”-shaped crypts (Fig. 22.7). They may contain dystrophic goblet cells with nuclei oriented toward the crypt lumen rather than toward the base. When evaluating studies of SSPs, one should be aware that there is high interobserver variability in the distinction between MVHP and SSP [42, 43]. According to a recent expert consensus panel, the presence of one or more crypts exhibiting the characteristic features of an SSP is sufficient to make the diagnosis [44].
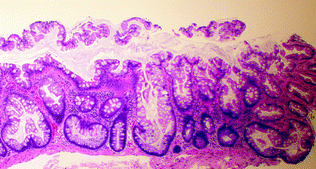
Fig. 22.7
Sessile serrated polyp
Although SSPs are considered non-dysplastic lesions, they may also give rise to dysplasia and carcinoma, and are considered precursor lesions to the serrated pathway of colorectal carcinogenesis [45]. Given that these polyps are more common in the right colon, and are more easily missed endoscopically or during pathologic evaluation, it is not surprising that screening colonoscopy has been much less effective at preventing right-sided (relative to left-sided) colorectal cancers [46, 47]. It has been theorized that the MVHP may be a precursor of SSP, since these two types of polyps share not only a morphologic resemblance, but also activating mutations in BRAF, a serine–threonine kinase involved in cell proliferation and anti-apoptotic pathways [48]. The development of dysplasia in SSPs has been linked to methylation of the MLH1 promoter, which results in microsatellite instability (MSI, see section on Molecular Pathology for details) [49]. It is thought that progression to invasive carcinoma occurs more rapidly in SSPs with dysplasia than in conventional adenomas [39, 50]. Currently, the exact risk and time interval of progression from an SSP to carcinoma is unknown.
22.5.2.3 Traditional Serrated Adenomas
The TSA is the least common type of serrated polyp, comprising <1 % of all serrated polyps, and is characterized by villiform architecture, tall, columnar epithelial cells with very eosinophilic cytoplasm, and ectopic crypt foci (small, horizontally oriented, bud-like crypts arising in the sides of villi that do not exhibit the usual anchorage to the muscularis mucosae) (Fig. 22.8). The nuclei of TSAs are ovoid or pencillate, with open chromatin and one or more small nucleoli, and do not exhibit the dense hyperchromasia or prominent nuclear stratification present in conventional adenomas. Unlike hyperplastic polyps and SSPs, TSAs are considered dysplastic lesions. Molecularly, TSAs appear to be a heterogeneous group. Based on their left-sided predominance and the presence of shared KRAS mutations, a subset of TSAs may arise from GCHPs [51, 52]. However, hyperplastic polyps and SSPs have also been proposed to be precursors to TSAs (particularly those found in the right colon), based on the presence of shared BRAF mutations [53]. Most carcinomas arising from TSAs are either microsatellite stable (MSS) or have a low degree of microsatellite instability (MSI-L) [54], and are associated with KRAS mutations and methylation of the MGMT gene [55].
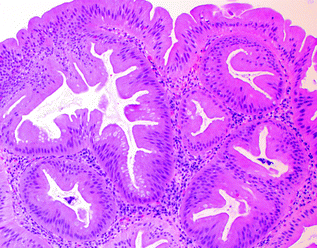
Fig. 22.8
Traditional serrated adenoma
22.5.2.4 Mixed and Unclassifiable Polyps
Although they do not comprise a formal category within the current WHO classification, serrated polyps that exhibit features consistent with more than one type of serrated polyp (mixed polyps, e.g., mixed SSP/TSA or unclassifiable polyps), or serrated polyp-like areas admixed with conventional adenomatous areas (mixed adenoma/TSA), do occur and are difficult to classify. These may reflect a yet-to-be-clarified biologic relationship between these polyp types.
22.5.3 Other Premalignant Lesions
Dysplasia of the conventional (intestinal) type may develop in two special polyp types, juvenile polyps and Peutz-Jeghers polyps, which are characteristic of their respective polyposis syndromes and will be described in the section on the genetics of hereditary colorectal cancer (see Sects. 22.7.5 and 22.7.6). Patients with IBD also have a well-recognized increased risk of colorectal carcinoma [56]. Dysplasia occurring in the setting of IBD has a special endoscopic-pathologic classification, which is beyond the scope of this chapter. For further information, the reader is referred to a number of excellent reviews on the subject [26, 57, 58].
22.6 Histopathology of Colorectal Cancer
Grossly and endoscopically, colorectal carcinoma may exhibit many different appearances, which are classified using the same Paris classification as dysplasia. Most carcinomas are sessile, centrally ulcerated lesions with irregular raised borders (Fig. 22.9). As they grow along the intestinal wall, they may involve the full circumference of the lumen, resulting in luminal narrowing and a classic “napkin ring” appearance. The cut surface of the tumor is firm, with a solid white to yellow-tan color. Mucinous cancers may have a villous surface covered by abundant mucin, with mucin oozing from pools in the cut surface of the tumor. Invasion beyond the muscularis propria is characterized by irregular areas of induration extending from the main mass into the surrounding pericolorectal fat, and peritoneal involvement is characterized by induration and puckering of the serosa overlying the involved segment of bowel.
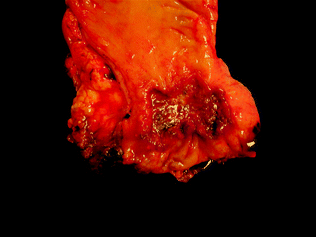
Fig. 22.9
Gross appearance of colorectal cancer. Image courtesy of Dr. Jeffrey C. Perumean, University of Texas, Southwestern Medical Center, Dallas, TX
Many histopathologic features have been assessed for colorectal carcinoma prognostication and prediction of response to therapy. Among the features with independent prognostic and/or predictive value in multivariate analyses that are frequently reported during pathologic evaluation are the histologic grade, stage, morphologic subtype, presence of lymphovascular and extramural venous invasion, peri- and intratumoral lymphocytic response, tumor budding, surgical margin status, and certain molecular markers.
22.6.1 Histologic Grading
The histologic grade of a colorectal tumor reflects the degree of differentiation (based on percentage gland formation) of the tumor and correlates inversely with prognosis. Traditionally, a three-tiered system has been used, where tumors with >95 % gland formation are well differentiated, those with 50–95 % gland formation are moderately differentiated, and those with <50 % gland formation are poorly differentiated. Due to high intra- and interobserver variability using this system, the American Joint Committee on Cancer (AJCC) has adopted a two-tiered system for evaluation of resected CRC specimens. Under this two-tiered system, well- and moderately differentiated tumors (which have been shown to have similar outcomes in most multivariate analyses) fall into the low grade category, whereas poorly differentiated tumors are considered high grade [9] (Fig. 22.10).
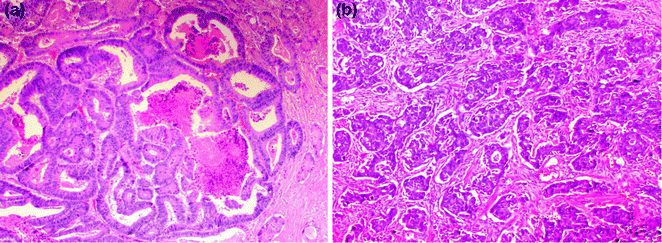
Fig. 22.10
Conventional adenocarcinoma. a Low grade. b High grade
22.6.2 Histologic Spread and Staging
The histologic stage is currently the most powerful predictor of CRC prognosis. The older Dukes’ staging system graded tumors from A to D. Using this staging system, stage A tumors are limited to the mucosa, stage B tumors reach into (B1) or through (B2) the muscularis propria, stage C tumors extend into (C1) or through (C2) the muscularis propria with positive lymph node involvement, and stage D tumors exhibit distant metastases [59]. The most widely used staging system today (TNM) is based on the depth of invasion into the bowel wall (T stage), as well as the presence or absence of metastasis to regional lymph nodes (N stage) and distant sites (M stage). For more detailed TNM staging information, as well as guidelines for the processing and evaluation of resected colorectal cancer specimens, the reader is referred to the most recent edition of the AJCC staging manual, as well as the College of American Pathologists’ protocol for the examination of these specimens [9, 60]. Although carcinoma is defined as any spread of neoplastic cells beyond the basement membrane, in the colon and rectum the risk of local and distant metastasis is close to zero for carcinoma that is restricted to the lamina propria or muscularis mucosae (intramucosal carcinoma) [9]. Therefore, intramucosal carcinoma is assigned the same T stage as high-grade dysplasia, and both are considered “in situ” lesions treatable by endoscopic excision, whereas the term “invasive carcinoma” is used to refer only to carcinoma invasive into or beyond the submucosa, which usually warrants surgical resection of the involved segment of colon or rectum.
Intraabdominal/pelvic spread of cancer occurs after perforation of the serosa. The first site of distant metastasis via a hematogenous route is the liver, via venous invasion. Extramural venous invasion portends the highest risk, and is an independent predictor of cancer recurrence and decreased survival [61]. The use of an elastic stain to highlight the elastic lamina, or a smooth muscle stain to highlight the muscular vessel wall, may be helpful in identifying vascular invasion in difficult cases. Lymphatic spread of a tumor occurs first to the pericolonic/perirectal lymph nodes, then to the mesenteric lymph nodes, and finally to the lungs and systemic circulation via the thoracic duct. Lymphovascular invasion (LVI) is identified by tumor emboli within endothelial-lined spaces (Fig. 22.11), sometimes surrounded by a layer of endothelial cells (“endothelial wrapping”). Endothelial markers, such as CD31 and CD34, may be used to stain endothelial cells, and the lymphatic-specific endothelial marker D2–40 may be used to highlight lymphatic invasion. Tumors may also spread via perineural invasion, which is characterized by tumor within the perineural sheath surrounding nerves.
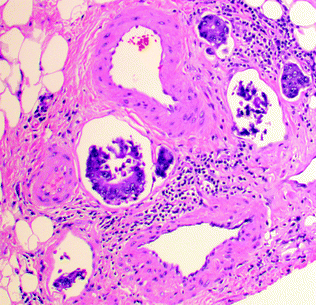
Fig. 22.11
Lymphovascular invasion by colorectal cancer
22.6.3 Histologic Subtypes
Many histologic subtypes of CRC exist, and an exhaustive discussion of all of these subtypes is beyond the scope of this chapter. Therefore, the following discussion pertains only to the most common subtypes, as well as those with established molecular pathologic relevance. For information on other subtypes, the reader is referred to the WHO Classification of Tumours of the Digestive System [62].
22.6.3.1 Conventional or “Usual” Type Adenocarcinoma
Conventional type adenocarcinoma is the most common histologic subtype of CRC. These account for 75–80 % of colorectal carcinomas, and are characterized by irregularly shaped, haphazardly arranged glands lined by tall columnar epithelial cells with variable amounts of intra- and extracellular mucin (Fig. 22.10). The glands may exhibit cribriform architecture and central inspissated mucus mixed with necrotic cellular debris (“dirty necrosis”). Poorly differentiated tumors may exhibit a predominantly solid growth pattern. In addition to the tall columnar enterocytes, tumors may contain variable numbers of goblet cells, Paneth cells, endocrine cells, and even benign squamous cells, melanocytes, and trophoblasts. Invasion of glands into the submucosa is typically accompanied by a stromal desmoplastic response characterized by a cellular, myxoid-appearing stroma containing plump myofibroblasts, collagen, and a variable inflammatory infiltrate, which surrounds the neoplastic glands in a streaming pattern.
22.6.3.2 Mucinous Adenocarcinoma
Mucinous, also known as colloid, adenocarcinomas represent 8–10 % of CRCs and are defined by WHO criteria as adenocarcinomas where >50 % of the tumor volume is occupied by extracellular mucin (tumors where the quantity of mucin is <50 % are diagnosed as adenocarcinomas with mucinous differentiation or mucinous features). These tumors are graded the same way as conventional carcinomas, where the low-grade end of the spectrum is represented by mucin pools lined by well-differentiated, tall, columnar epithelium, and the high-grade end is represented by poorly differentiated and/or signet ring cells floating within mucin pools (Fig. 22.12). With the exception of those that have a high degree of microsatellite instability (MSI-H), mucinous carcinomas tend to present at more advanced stages, and have also been associated with a worse prognosis, independent of stage and other prognostic factors [63, 64]. The molecular basis for the mucinous phenotype has been linked to the expression of the intestinal epithelial transcription factor, MATH1, which activates MUC2 expression [65]. In addition, the transforming growth factor beta (TGFβ) pathway has been associated with the mucinous phenotype. SMAD4 mutation is associated with mucinous CRC in humans [66] and mice [67], and loss of Tgfbr2 and Pten results in development of mucinous adenocarcinoma in genetically engineered mice [68].
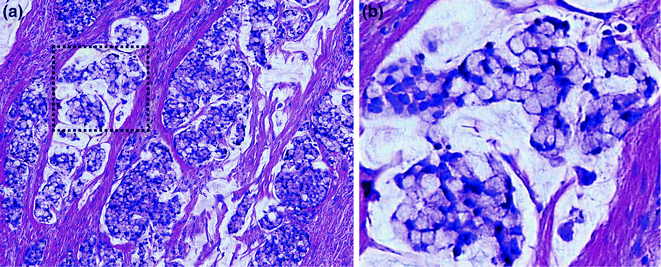
Fig. 22.12
Mucinous adenocarcinoma with signet ring cells. a Low magnification. b High magnification of region outlined in panel A
22.6.3.3 Signet Ring Cell Adenocarcinoma
Signet ring cell adenocarcinoma comprises up to 1 % of all CRCs, and is defined by the presence of at least 50 % signet ring cells within the tumor. Grossly, it may appear to be a well-defined mass or, if diffusely infiltrative, an ill-defined area of mural thickening or stricturing. Microscopically, it is characterized by single infiltrating cells and clusters of cells, often with a large cytoplasmic vacuole containing mucin or other cytoplasmic contents, which gives the cell a round-to-ovoid shape and indents the nucleus, forming a profile that resembles a signet ring (Fig. 22.12). When an adenocarcinoma contains >50 % extracellular mucin and signet ring cells, it should be classified as a signet ring cell carcinoma. By definition, signet ring cell carcinoma is considered high grade, and, except when MSI-H, has been associated with a poor prognosis, with a 5-year survival rate of <10 % [69, 70].
22.6.3.4 Medullary Carcinoma
Also referred to as lymphoepithelioma-like carcinoma and large cell minimally differentiated carcinoma, medullary carcinoma is the subtype most classically associated with MSI-H, and may occur in the setting of both hereditary (Lynch syndrome, usually with MSH2 mutations) and sporadic colorectal cancer (MLH1 promoter methylation) [71]. Medullary carcinomas are associated with right-sided location, female gender (usually older females), lower incidence of lymph node metastasis, and typically have a good prognosis [72–74]. Morphologically, these are poorly differentiated or undifferentiated tumors composed of solid sheets, nests, or trabeculae of polygonal tumor cells with vesicular chromatin, prominent nucleoli, and abundant cytoplasm with a syncytial appearance. These are associated with a prominent peritumoral and intratumoral lymphocytic infiltrate [72] (Fig. 22.13). These tumors are well circumscribed, with a broad-front pushing (as opposed to an infiltrative) growth pattern. Unlike typical colorectal carcinomas, which are CK7−, CK20+, and CDX-2+ by immunohistochemistry, medullary carcinomas tend to be CK7 variably positive, CK20−, and CDX-2– or only weakly positive [71], and are positive for the mesothelial marker Calretinin [74].
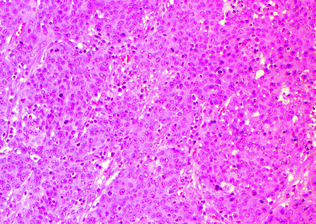
Fig. 22.13
Medullary carcinoma
22.6.3.5 MSI-H Adenocarcinoma
Approximately 10–15 % of colorectal cancers are MSI-H [75], and, although these tumors do not comprise a single distinct morphologic subtype, they nonetheless have been shown to share some common histopathologic features. MSI-H CRC may be any one of the previously discussed morphologic subtypes (conventional, mucinous, medullary, and signet ring cell), are frequently right-sided, polypoid, exophytic masses that lack the “dirty necrosis” typical of most colorectal carcinomas, and have in common the presence of tumor-infiltrating lymphocytes (TILs), the most sensitive and specific morphologic marker, to date, of MSI-H status [76–78]. TILs are CD3+ CD8+ cytotoxic T cells that infiltrate between epithelial cells within the tumor. According to the current (2016 edition) CAP criteria, a colorectal carcinoma is positive for TILs if it contains at least three TILs per high-power field (HPF). Other features that have been associated with MSI-H status include a peritumoral lymphocytic infiltrate, which surrounds, but does not infiltrate, malignant glands and cell clusters, and may form nodular aggregates at the deep periphery of the tumor, resembling the lymphoid aggregates seen in Crohn’s disease (peritumoral “Crohn’s-like” infiltrate). Poor tumor differentiation and a mucinous phenotype have also been associated with MSI-H tumors [79, 80].
As an aside, tumors with a CpG island methylation phenotype (CIMP+, see Molecular Pathology section for details) may be either MSI-H or not. Those that are MSI-H share the same features as other MSI-H tumors. Those that are not MSI-H are more often right-sided and poorly differentiated but, unlike MSI-H tumors, do exhibit “dirty necrosis” and tend to lack TILs or a peritumoral lymphocytic response [81].
As a group, patients with MSI-H tumors have better overall survival compared to non-MSI-H tumors, when controlled for stage as well as multiple other prognostic factors [82–85]. This may in part reflect a stronger antitumor immune response, as evidenced by the prominent peri- and intratumoral lymphocytic infiltrates seen in these tumors. Because the commonly used chemotherapeutic agents 5-FU and oxaliplatin require a functional DNA mismatch repair (MMR) system in order to promote tumor cell apoptosis, MSI-H tumors are less responsive to these agents, but have been shown to be sensitive to the topoisomerase inhibitor irinotecan, which induces double-stranded DNA breaks. In fact, current National Comprehensive Cancer Network guidelines state that adjuvant 5-FU therapy should not be offered to stage II colorectal cancer patients with MSI-H cancers, as it is not only of little additional benefit (since these patients have a very good prognosis to begin with), but may also cause harm [86].
22.6.3.6 Serrated Adenocarcinoma
“Serrated adenocarcinoma” is not a distinct subtype in the current WHO classification, and has been used to refer to both adenocarcinomas with a serrated morphology and adenocarcinomas arising via the serrated pathway of carcinogenesis. Morphologically, these tumors often exhibit a serrated luminal contour to their glands and frequently produce mucin. Epithelial cells within these tumors are characterized by eosinophilic or clear cytoplasm, and vesicular, ovoid nuclei with a peripheral rim of condensed chromatin (Fig. 22.14). Serrated carcinomas usually lack the “dirty necrosis” of conventional colorectal carcinoma [87, 88]. Clinicopathologically and molecularly, these comprise at least two distinct subgroups: those that are MSI-H right-sided cancers arising in SSPs, and those that are MSS/MSI-L left-sided cancers arising in TSAs [87].
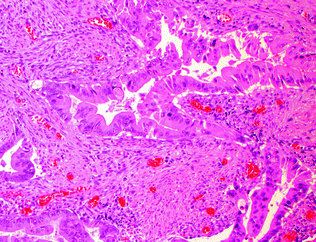
Fig. 22.14
Serrated adenocarcinoma
22.6.4 Other Prognostic Histopathologic Markers
Tumor budding, which is thought to represent dedifferentiation to a more stem cell-like phenotype (often referred to as epithelial–mesenchymal transition), has been independently associated with a worse prognosis [89]. Although this is currently a controversial area due to the lack of standardized criteria or grading systems, the generally accepted definition of tumor budding is the presence of single epithelial tumor cells or clusters of <5 cells lacking well-formed glandular architecture, which are present at the deep invasive front of a tumor and associated with a characteristic desmoplastic reaction (Fig. 22.15) [90]. There is currently no standardized threshold for the degree of budding that is prognostically significant, with one study defining high-grade tumor budding as the presence of >1 bud in 5 (20x) HPF, or >10 tumor buds in 10 HPF [91]. Regardless, the presence of tumor budding has become increasingly incorporated as a routine component in pathology reports.
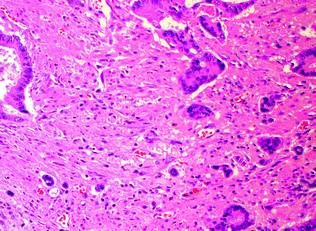
Fig. 22.15
Tumor budding
The antitumor immune response, particularly the lymphocyte-mediated component, has gained prominence in recent years due to its association with MSI-H colorectal cancer and an improved prognosis in multivariate analyses [92, 93]. Components of the lymphocytic antitumor immune response include TILs, peritumoral lymphocytes and lymphoid aggregates, and reactive regional lymph nodes. It has been hypothesized that, in MSI-H tumors, the T-cell mediated response is directed against frameshift-induced neopeptides [94]. Although several grading systems for the degree of antitumor lymphocytic response have been proposed, there is currently no standardized, clinically validated grading system, although one will likely emerge in the future.
The concept of cancer-initiating stem cells, which are characterized by the ability to self-renew and differentiate into multi-lineage progenitors, has gained prominence over the last two decades due to their observed resistance to therapy and purported role as the cells that initiate and drive tumorigenesis and recurrence [95–97]. Although several potential stem cell markers have been identified in colorectal cancer (CD133, LGR5, CD166, CD24, MSI-1, BMI, Aldehyde dehydrogenase, and DCM kinase-like II) [98], none of these currently have established value as prognostic markers or therapeutic targets, and this is an area requiring further investigation.
22.7 Genetics of Hereditary Colorectal Cancer
A number of hereditary syndromes are associated with an increased risk of CRC and will be discussed in the following section. Two other syndromes, Cowden syndrome and Cronkhite–Canada syndrome, are associated with the development of colorectal polyps; for a discussion of the former, the reader is referred to the chapters on ovarian/uterine and breast cancer. The latter syndrome does not have an established association with CRC, and will not be discussed further.
22.7.1 Familial Adenomatous Polyposis (FAP) and Variants
One of the best-known familial CRC syndromes is familial adenomatous polyposis (FAP), an autosomal dominant condition characterized by germline mutations in the Adenomatous Polyposis Coli (APC) gene on chromosome 5q. Over 800 disease-associated mutations in APC have been identified, >90 % of which are nonsense or frameshift mutations leading to a truncated protein product [99]. Many of these mutations have specific phenotypic correlations. Patients develop hundreds (and usually thousands) of colorectal adenomas within the first two decades of life that eventually progress to CRC (on average, by age 45) unless the entire colorectum is prophylactically resected. Additionally, patients with FAP may develop a number of extracolorectal manifestations, including desmoid tumors, small intestinal adenomas, dysplastic gastric fundic gland polyps, jaw osteomas, congenital hypertrophy of the retinal pigment epithelium (CHRPE), epidermal cysts, multiple endocrine neoplasia (MEN), pancreatic adenocarcinoma, hepatoblastoma, extrahepatic cholangiocarcinoma, and papillary thyroid carcinoma [100]. Gardner’s syndrome is an FAP variant with extraintestinal manifestations, particularly desmoid tumors and osteomas. One of the two variants of Turcot’s syndrome is another variant of FAP, where patients also present with medulloblastoma. Attenuated FAP (AFAP) is a milder variant of FAP characterized by the presence of <100 adenomas, a milder phenotype, and later clinical presentation.
22.7.2 MUTYH-Associated Polyposis (MAP)
MUTYH-associated polyposis (MAP) is an autosomal recessive colorectal polyposis syndrome characterized by biallelic germline mutations in the MUTYH gene on chromosome 1p34.1, which encodes a glycosylase involved in DNA repair after oxidative damage [101]. Defective MUTYH function causes an increased rate of G > T transversions in tumor suppressors and oncogenes, with the most common target being APC. Patients have a variable number of polyps (including adenomas and serrated polyps) and an increased risk of CRC. The most common phenotype resembles that seen in AFAP patients, with most patients having <100 polyps, some having >100, and a few having >1000 polyps. The average age at presentation is 45 years [102, 103], and 60 % of patients have CRC at initial presentation [104]. MAP may account for up to 42 % of patients with adenomatous polyposis who do not have APC mutations [105].
22.7.3 Lynch Syndrome
Lynch syndrome is an autosomal dominant cancer syndrome caused by germline mutations in one of four DNA MMR genes (MLH1, PMS2, MSH2, and MSH6) (discussed in more detail in Sect. 22.13). In addition, EPCAM mutations have been implicated in Lynch syndrome, as they lead to transcriptional silencing of MSH2 via promoter methylation [106]. Rarely, heritable epigenetic silencing of MLH1 or MSH2 may occur [107–109]. Lynch syndrome accounts for 10–25 % of familial colorectal cancer and 3 % of all colorectal carcinomas [110]. The vast majority of patients (~90 %) have mutations in MLH1 or MSH2 [111–113], with PMS2 and MSH6 mutations being much rarer. Mutations in another MMR gene, MSH3, have also been documented in the setting of MLH1 deficiency, but to date, no germline mutations in MSH3 have been found [114, 115]. In total, over 1000 different mutations in the MMR genes have been identified [116].
The term hereditary nonpolyposis colorectal cancer (HNPCC) is often used synonymously with Lynch syndrome, which is not entirely accurate on two counts. First, HNPCC is a clinical designation for patients with familial colorectal cancer predisposition meeting the Amsterdam criteria [117], and actually encompasses two patient groups: those with Lynch syndrome and those of currently unknown genetic cause, referred to as Familial Colorectal Cancer Type X [118]. Second, Lynch syndrome patients do also develop colonic polyps, albeit not as many as in the adenomatous polyposis syndromes (usually <15 polyps).
The lifetime risk of CRC with Lynch syndrome ranges from 10 to 53 %, depending on the MMR gene that is mutated [119], with the average age at development of CRC being 40–50 years. In approximately 20 % of patients, multiple CRCs may develop, either in a synchronous or metachronous fashion. Most Lynch syndrome patients are born with a mutation in one allele, with the second allele lost by somatic mutation, loss of heterozygosity, or epigenetic silencing. Rare patients are born with biallelic mutations and are characterized by pediatric CRC, as well as other pediatric hematologic and brain malignancies [120–122].
In addition to the increased risk of CRC, Lynch syndrome patients are also at increased risk for endometrial carcinoma (15–44 %), ovarian carcinoma, gastric and small intestinal adenocarcinoma, carcinomas of the renal pelvis and ureter, hepatocellular carcinoma, cholangiocarcinoma, sebaceous tumors, and brain tumors, among others [123–126]. Lynch syndrome patients with primary brain tumors (often glioblastoma) comprise one of the two subtypes of Turcot syndrome (the other being FAP patients with medulloblastoma). Muir–Torre syndrome is a variant of Lynch syndrome characterized by the presence of sebaceous tumors and keratoacanthomas.
22.7.3.1 Routine Testing for Lynch Syndrome in Colorectal Cancer
Although there are well-established clinical criteria for identifying patients at risk for Lynch syndrome (Amsterdam II criteria [127] and the Revised Bethesda Criteria [128]), they will miss a significant number of patients [129–131]. Furthermore, although Lynch-associated CRCs often exhibit morphologic features of MSI-H tumors, none of these features are helpful in distinguishing Lynch-associated from sporadic MSI-H tumors. Therefore, the primary screening method for the detection of Lynch syndrome in CRC patients is immunohistochemistry for protein expression of components of the MMR complex. This method is >90 % sensitive and nearly 100 % specific for identifying MMR deficiency when one or more of the four MMR proteins (MLH1, PMS2, MSH2, and MSH6) are lost [132, 133]. Immunohistochemistry may rarely miss some mutations that lead to loss of function without loss of immunoreactivity (for example, when the mutation occurs away from the epitope detected by the antibody).
As MLH1/PMS2 and MSH2/MSH6 function as dimer pairs, loss of immunostaining of the dominant partner, MLH1 or MSH2, results in concurrent loss of immunostaining of the nondominant partner, PMS2 or MSH6. Conversely, when PMS2 or MSH6 are lost, there is still some retained immunostaining for MLH1 and MSH2, because the latter two proteins may form dimers with other partners, such as MLH3 [134, 135]. In most cases, the background stromal and inflammatory cells or adjacent normal crypts retain MMR protein expression, serving as positive controls when determining whether there is MMR protein loss in tumor cells (Fig. 22.16). This occurs because the allele carrying the wild-type MMR gene is only inactivated in tumor cells, allowing detection of the wild-type protein in surrounding normal tissue where the unaffected allele is still intact. An exception occurs when a patient has biallelic germline mutations, which result in loss of immunostaining in normal tissues as well.
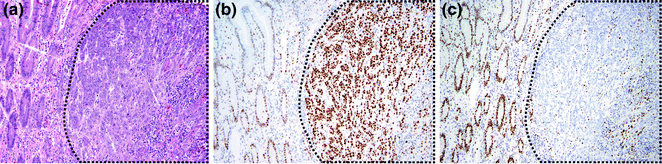
Fig. 22.16
Mismatch repair immunohistochemistry. a H&E staining of the border between normal colon and CRC. b Immunohistochemistry for MLH1. Both tumor cells and normal cells stain positively. c Immunohistochemistry for MSH2. Normal cells are positive, but tumor cells are negative. Positively staining cells within the tumor mass are non-epithelial cells. In all panels, the cancer is outlined in black
MLH1 deficiency may be due to either Lynch syndrome or MLH1 promoter methylation, so loss of MLH1 expression by immunohistochemistry may be followed up by MLH1 promoter methylation testing by PCR, or germline testing for MLH1 mutations. Because the BRAF V600E mutation is often associated with MLH1 methylation in sporadic colorectal cancers, but almost never present in Lynch-associated colorectal cancers, testing for this mutation may be helpful in distinguishing sporadic from Lynch-associated tumors with MLH1 deficiency [136]. When MMR immunohistochemistry is inconclusive or is positive for all four MMR proteins in a patient with high clinical suspicion for Lynch syndrome, MSI testing by PCR or germline testing for MMR mutations may be useful. Current guidelines recommend testing all newly diagnosed colorectal cancers for Lynch syndrome by MMR immunohistochemistry, MSI PCR, or both of these methods [137, 138].
22.7.4 Serrated Polyposis Syndrome (SPS)
Serrated polyposis syndrome (SPS) is a hereditary CRC syndrome of unknown molecular etiology, characterized by the presence of increased numbers of serrated polyps (either hyperplastic polyps or SSPs) and an estimated lifetime risk of developing colorectal cancer of 50 % (97). Two subtypes have been identified: Type 1 is characterized by mostly hyperplastic polyps and a low risk of CRC, while Type 2 is characterized by mostly SSPs, with or without hyperplastic polyps, and a higher risk of CRC. Approximately 33–53 % of colorectal cancers in SPS patients are thought to arise via the CIMP pathway [139–141]. Conventional adenomas may also be present in >80 % of SPS patients.
22.7.5 Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome (PJS) is an autosomal dominant syndrome characterized by mucocutaneous pigmentation and GI tract polyps with a unique arborizing smooth muscle network. These Peutz-Jeghers polyps usually present within the first two decades of life (Fig. 22.17). Patients have an increased risk of carcinoma of the colorectum, small intestine, stomach, pancreas, esophagus, ovary, uterus, lung, and breast [142]. The syndrome is associated with germline mutations in LKB1 (also called STK11), a tumor suppressor gene encoding a serine–threonine kinase that modulates cellular energy pathways and maintains cell polarity [143]. About 15–30 % of Peutz–Jeghers syndrome patients develop dysplastic polyps and carcinoma [144, 145]. Rare occurrences of solitary sporadic Peutz–Jeghers polyps have been reported. The clinical significance of these lesions remains unclear, but removal is recommended, as these polyps may harbor dysplasia [146, 147].
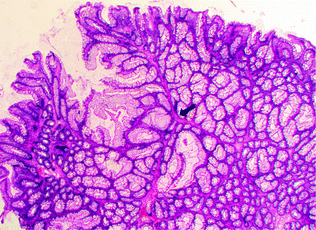
Fig. 22.17
Peutz-Jeghers polyp
22.7.6 Juvenile Polyposis Syndrome (JPS)
Juvenile polyposis syndrome (JPS) is an autosomal dominant GI polyposis syndrome characterized by the presence of multiple juvenile polyps within the colorectum and proximal GI tract, including the stomach. JPS is most commonly associated with mutations in the TGFβ signaling pathway, including germline mutations in SMAD4 (15 %) and Bone Morphogenetic Protein Receptor Type 1A (BMPR1A; 25 %), which occur in 40–60 % of JPS [148, 149]. A small percentage of JPS patients (5 %) have mutations in PTEN, a negative regulator of AKT signaling [150, 151]. Juvenile polyps are a morphologically distinct type of non-neoplastic mucosal polyp characterized by disorganized, cystically dilated glands filled with mucus, surrounded by an edematous, inflamed stroma (Fig. 22.18). Although juvenile polyps may occur sporadically in children, and as solitary sporadic polyps in adults, there is no increased risk of carcinoma unless the polyps occur in the syndromic setting (for detailed clinical criteria for the diagnosis of JPS, please refer to [152]). In syndromic patients, there is an increased risk of colorectal, gastric, and pancreatic carcinoma, with an estimated 17–22 % risk of colorectal cancer by age 35 years, which increases to 68 % by age 60 [142]. Carcinogenesis occurs by the adenoma-carcinoma pathway in juvenile polyps that develop conventional dysplasia.
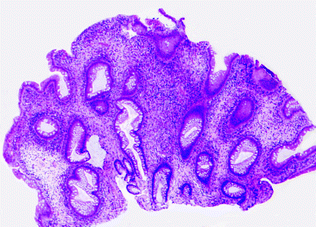
Fig. 22.18
Juvenile polyp
22.7.7 Mixed Polyposis Syndrome
Mixed polyposis syndrome is an autosomal dominant syndrome characterized by the presence of “mixed” polyps with adenomatous, juvenile, and hyperplastic features, and an increased risk of CRC [153]. It is currently unclear whether this represents a variant of juvenile polyposis syndrome or serrated polyposis syndrome, but associations with BMPR1A and GREM-1 mutations have been described [154–156].
22.8 The Molecular Pathology of Colorectal Cancer
CRC presents as sporadic, inherited, or familial disease. Sporadic disease, in which there is no family history, accounts for approximately 70 % of all CRC cases. This pattern of disease derives from stochastic somatic mutations that are driven by both dietary and environmental risk factors. Inherited disease (described above) accounts for only 5 % of CRC cases and typically presents with well-characterized predisposing mutations. These cases are subdivided based on whether the patients display colonic polyps, as in familial adenomatous polyposis (FAP) or MUTYH-associated polyposis (MAP), or those without polyps (Lynch syndrome). The third, and least understood, pattern is familial CRC, which accounts for 25 % of cases. Affected patients have a family history of CRC, similar to patients with inherited disease, but the genetic etiology has not been defined. The molecular pathogenesis of CRC has been extensively studied and will be described in detail in this chapter. CRC can develop via one of two major molecular pathways of tumorigenesis: CIN or MSI. These CRC subclasses have different mutational spectra and are therefore likely to require distinct therapeutic strategies as targeted therapies become available.
22.9 Initiation of CRC Tumorigenesis
Specific genetic mutations are thought to drive the transformation from normal colonic epithelium to invasive cancer. In 1990, Fearon and Vogelstein described a multistep genetic model of colorectal carcinogenesis in which each accumulated genetic alteration contributed to the progression from normal epithelium to adenoma to invasive cancer (Fig. 22.19) [157]. Although not the complete story, this model has become the basis for much of our understanding of how CRC tumorigenesis progresses. It was quickly observed that cancer cells contained a high incidence of mutations compared to normal cells, which contained very few mutations. This observation led to the idea that one of the initial steps in carcinogenesis occurs when cancer cells acquire mutations in genes that maintain genomic instability, which facilitates an increased mutation rate and thereby, a “mutator phenotype” [158]. In contrast, a second theory suggests that tumor initiation depends less upon increased mutational rate than on positive clonal selection driven by an advantageous mutation [159]. In this case, a normal mutation rate is sufficient for tumor initiation if positive selection is accounted for [159, 160]. Recent work suggests that since normal somatic cells acquire mutations at the same rate [161], the risk of cancer developing in an organ is associated with the number of lifetime stem cell divisions that occur within a given tissue [162]. Based on this theory, mutations accumulate randomly by chance during DNA replication. Since the colonic epithelium is continuously turning over, the risk of mutation in this tissue is high compared to less proliferative tissues. It is estimated that the time between adenoma initiation and carcinoma development in the colon is about 17 years, while the time between carcinoma and metastasis is only 1.8 years [161]. These estimates suggest that for adenoma to progress, it has to acquire more mutations and undergo more clonal expansions than a carcinoma that metastasizes. Based on this model, it is likely that most, if not all, of the mutations required for metastasis are present in the parent carcinoma.
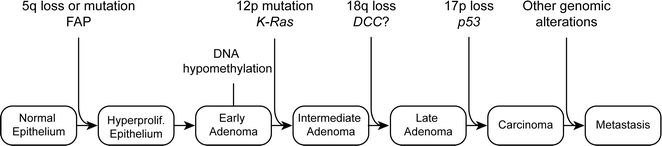
Fig. 22.19
Original genetic model of CRC development as proposed by Fearon and Vogelstein in 1990. Adapted from Fearon and Vogelstein [163]
22.10 Mechanisms Leading to Chromosomal Instability (CIN)
CRCs can be divided into classes based on the general state of the genome. The majority (~70 %) of sporadic CRCs contain a high frequency of gain and/or loss of whole chromosomes or chromosomal segments. This phenotype is known as chromosomal instability (CIN) and is characterized by chromosomal translocations, amplifications, deletions, and insertions [164, 165]. One of the underlying causes of such aneuploidy is improper segregation of chromosomes. The mitotic spindle checkpoint maintains fidelity of chromosome segregation by delaying anaphase onset until chromosomes are properly aligned on the mitotic spindle [166]. A second mechanism of aneuploidy is driven by centrosomes, which secure cytoplasmic microtubules to the mitotic spindle during cell division. Abnormal centrosome number results in mulitpolar spindles, ultimately causing chromosomal missegregation [166]. Genomic integrity is also maintained by a second checkpoint, the DNA damage checkpoint, which protects cells against postreplicative DNA damage and genotoxic stress. Failed DNA damage checkpoints allow damaged DNA to progress through mitosis, which can result in chromosomal structural alterations [164]. Loss of heterozygosity (LOH) is one of the primary outcomes of chromosomal instability, and CRC tumors may display up to 40 % of parental allelic loss [164, 167]. The most frequent CRC-associated chromosomal losses occur at 1p, 4q, 5q, 8p, 14q, 15q, 17p and q, 18p and q, 20p, and 22q [168]. Of note, loss of 18p and q (containing SMAD4) or 17p and q (containing TP53), the most common chromosomal deletions, occur in up to 66 and 56 % of CRC, respectively [168]. The exact mechanisms that underlie LOH are not well understood, but several pathways have been implicated, including mitotic nondisjunction, homologous recombination, and chromosomal structural changes originating from recombinations and deletions following DNA double strand break repair [164, 169].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


