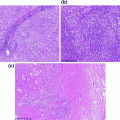
Fig. 16.1
Histological features of normal cell types within the brain. a Arachnoidal collection of meningeal cells and psammomatous calcification similar to those seen in meningiomas, b oligodendrocytes within white matter with perinuclear halos analogous to those seen in oligodendrogliomas, c reactive astrocytes with eosinophilic cytoplasm similar to that in astrocytomas
The brain parenchyma proper is segmented into lobes which each can be affected by all major primary brain tumor types. However, the frequency of each tumor type can vary significantly based on location and the relative abundance of particular cell lineages. Each lobe contains a similar overall architecture with an outermost gray matter layer that consists of a cortical ribbon containing a large number of neurons relative to glia. Below this layer is the white matter that consists of a vast number of glia including myelinating oligodendrocytes and astrocytes (Fig. 16.1b and c, respectively). Throughout the brain, the NG2-cells represent the largest mitotically active cell population within the brain, and are OLIG2-expressing oligodendroglial progenitors [11]. The white matter serves as a “superhighway” of axons but very few neuronal bodies and is more abundant than the gray matter overall. The different incidence of cell types and different progenitor types within each region of the brain is thought to be the basis for differences where each brain tumor most commonly arises. Oligodendrogliomas arise most frequently in regions of the frontal lobes where white matter is most abundant. Astrocytomas or glioblastomas are felt to arise more frequently from deeper locations around the ventricles presumably closer to where stem cells of the brain reside. Such patterns have recently also been supported based on imaging studies of brain tumor growth patterns [12].
The brainstem and cerebellum are some of the most primitive and basic functional regions of the brain, and together reside in the area of the cranium termed the posterior fossa. While adult primary brain tumors rarely arise in either of these regions, these structures are one of the most frequent sites of primary brain tumors within children. This is possibly due to the normally late stem and progenitor cell development that occurs in these structures [13]. The spinal cord extends for most of the length of the spinal canal and contains similar anatomic structures and cell types of neurons, astrocytes, and oligodendrocytes as is seen in other regions of the brain. The spinal cord is an infrequent site for tumors in adults and children although several specialized types of tumors are known to occur there, some almost exclusively (e.g., myxopapillary ependymoma).
16.4 General Neuropathology Principles
16.4.1 Brain Tumor Classification
Brain tumors were first classified based on the resemblance of their microscopic cell morphology to normal mature cell types and structures within the brain (Fig. 16.1). The first widely employed classification system was established in this manner by Bailey and Cushing who were particularly struck by the similarities of brain tumor cells to normal differentiated cells and undifferentiated progenitor cells of the developing brain [14]. The names most commonly used today still derive from their conventions with the “blastoma” terminology a reminder of the original developmental perspective. This strategy for classification was a useful one for many years, and as the development of the brain and its tumors has become better understood it is even more clear that the tumors resemble earlier stages of developing progenitor or neural stem cells more than they have analogy to differentiated cell types [15].
Histomorphologic assessment remains the most widely used biomarker classification of brain tumors in clinical practice. This is one of the most important functions performed by pathologists in their care of patients with important distinctions made by the initial assignment of tumors as gliomas versus other tumor types. This assessment is based on microscopic examination of hematoxylin and eosin (H&E) staining which is the standard approach to the pathologic evaluation of most cancers. Tumors are first assessed for the class of the tumor that is commonly referred to as the diagnosis (Fig. 16.2). Some of the most common diagnoses or classes of primary brain tumors are gliomas (glioblastoma, astrocytoma) or meningiomas but the range of diagnostic categories is extremely broad and there are currently more than 100 distinct brain tumor types recognized by the WHO classification schema [16]. Many of these are rare tumors with distinct appearances and biology associated with them, particularly those that occur in children.
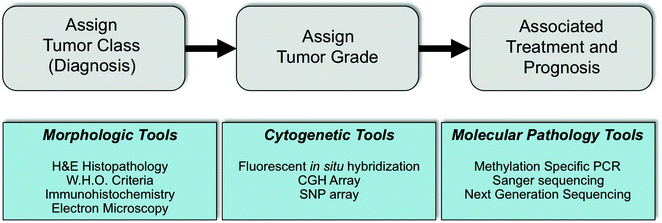
Fig. 16.2
Diagnostic approach and tools used in neuropathology practice. Gray bubbles indicate the workflow of assigning brain tumor class and grade by morphologic assessment. Such assignments lead to information on treatment and prognosis for individual patients. In blue are the clinical tools commonly utilized in modern pathology practice to aid in classification and prognostic assignment of patients and their tumors
16.4.2 Brain Tumor Grading
Once a class of tumor lineage has been specified, then the associated grading criteria are applied to the tumor. As such the grade can only be accurately assigned once a tumor class or classes have been decided upon (Fig. 16.2). Grading criteria are most commonly based on the degree of cellularity, atypia, nuclear size relative to the cytoplasm, and degree of proliferative or mitotic activity. The higher the histologic grade, then the more aggressive the tumor is generally and with a less favorable prognosis implied with respect to recurrence risk or survival metrics. The WHO grading assignments are from Grade I to Grade IV with the higher the number the more aggressive the tumor appears. Tumors sometimes contain more than one grade type, and in these instances, the highest grade determines the overall grade of the tumor, even if most of the tumor is made up of low-grade cells. While some general categories of tumors are felt to progress with recurrence or time to a higher histologic grade as part of their natural progression, (e.g., Grade II diffuse astrocytoma progressing to a Grade III anaplastic astrocytoma) most tumors do not have more than one associated grade or show progression. The clinical course of each grade can be varied but generally WHO Grade I brain tumors are slow growing and often curable by surgical resection alone if completely excised, while Grade IV lesions grow rapidly and are fatal diseases if not treated.
16.4.3 Brain Tumor Staging
One of the more unique aspects of brain and spinal cord tumors is that they rarely spread or metastasize to other parts of the body. This is hypothesized to be due in part to the blood–brain barrier or other specialized aspects of the brain vasculature. However, circulating GBM and other tumor cells have recently been described in peripheral blood, albeit at much lower levels than seen in other cancers. As a result of their lack of spread, there is no formal staging system that is used in clinical practice. However, CNS tumors are capable of spreading within the central nervous system and affecting other parts of the brain and spinal cord. This is most often felt to occur through sloughing of cells into the cerebrospinal fluid and “seeding” most typically occurs in the spinal region due to gravity and positional factors. This form of staging is usually assessed by examination of cerebrospinal fluid or on radiologic imaging. Another important aspect of evaluation of brain tumors is imaging taken after surgery to assess the extent of resection of a tumor. While the most common tumors of the CNS cannot be fully removed due to their diffuse infiltration of normal structures (e.g., diffuse astrocytoma, glioblastoma) the extent of resection of the tumor and amount left behind can affect the patient’s outcome significantly in certain tumor types. Treatment planning is also commonly affected by whether a tumor is completely resected (gross total resection) or subtotally resected.
16.5 Pathology of Brain Tumors
Given the breadth of brain tumor types, we will only cover the most common tumors and grades encountered in practice for meningioma, oligodendrogliomas, astrocytomas, and glioblastomas, each described separately below.
16.5.1 Meningioma
Meningiomas are one of the most common intracranial tumors and represent approximately 30 % of brain tumors in the US and occur almost exclusively in adults [16]. Generally, such tumors are slow growing and benign and may be discovered incidentally on MRI or at autopsy. These tumors can arise in any location where there are meningeal coverings to the brain. Occasional tumors may arise in the choroid plexus or spinal nerve root location causing some diagnostic confusion if not carefully assessed. The most common sites are the convexity of the brain (falx, parasagittal regions) and skull base regions (olfactory region, sphenoid, suprasellar).
Meningiomas are grossly well circumscribed and tend to push aside the brain parenchyma without invading or destroying it. They are most commonly attached to the inner surface of the dura and sometimes erode the overlying bone of the skull. The tumors rarely spread within the intracranial compartment or to other sites in the body. They typically occur singly but when present as multiple lesions these can occur sporadically or in the setting of neurofibromatosis type 2 (NF2) syndrome.
Currently, the WHO recognizes 15 distinct histological subclasses of meningioma with the most common being WHO Grade I tumors such as meningothelial, fibrous, or transitional classes (Fig. 16.3a). Each have a strong morphologic resemblance to the normal arachnoidal meningothelial cells seen within the dura and leptomeninges (Fig. 16.1a). Other WHO Grade I subtypes include psammomatous, angiomatous, microcystic, secretory, lymphoplasmacyte-rich, and metaplastic meningiomas. Grade II tumors have increased proliferative rate, cellularity, atypia, or nuclear cytoplasmic ratio, or loss of a normal morphologic pattern. Several specific subclasses are also automatically categorized as Grade II due to an increased recurrence risk including chordoid and clear cell variants; tumors with further evidence of mitotic activity, necrosis, and near complete loss of meningiomatous differentiation. Specific variants, namely the anaplastic, papillary, and rhabdoid meningiomas are also assessed WHO Grade III due to their strong recurrence risk. The presence of brain invasion is considered an independent recurrence risk sign.
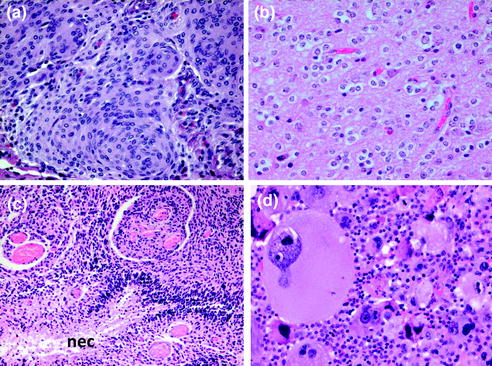
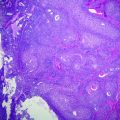
Fig. 16.3
Histological features of common brain tumors a meningioma with typical whorls, b oligodendroglioma with diffuse infiltrating cells surrounded by perinuclear halos, c glioblastoma with pseudopalisading necrosis (nec) and dense atypical tumor cells, d glioblastoma with markedly atypical giant cells
Meningiomas are often recognized by their H&E morphology, however, in cases where the diagnosis is in question this can be aided by use of immunohistochemistry (IHC). The most common positive identification marker expressed in meningiomas is epithelial membrane antigen (EMA) [17]. The expression of this marker, however, is often weak and diffuse and therefore not always reliable for identification. This is particularly true in that higher grade meningiomas lose the expression of this marker. Given their origin from non-neuroepithelial cell types, meningiomas lack expression of primary brain tumor markers such as the glial markers OLIG2 or GFAP which also may be helpful in their distinction from these entities. Electron microscopy is occasionally used to identify key features of the mesenchymal phenotype when other markers may have failed. Future studies to identify specific markers of the lineage for meningeal cells in addition to EMA are needed and would aid in the diagnostic evaluation of these tumors. Further study of the complex developmental origins of the normal meningeal cells and the different cell types in the brain’s coverings will likely lead to such discoveries as they have in other tumors.
16.5.2 Oligodendroglioma
Oligodendrogliomas are rare diffusely infiltrating tumors of adults that have a very distinctive histological appearance on H&E staining and a similar appearance to normal differentiated oligodendrocytes of the brain. The tumor cells are highly uniform with rounded nuclei and a monomorphic or “clonal” appearance overall. Their most distinctive feature is the presence of clear spaces in formalin-fixed paraffin-embedded (FFPE) sections around each cell which is described as a perinuclear halo or the “fried egg” appearance (Fig. 16.3b). The nucleus in oligodendroglioma cells is very centrally located within the halo and is present in abundance throughout the tumor. This should be distinguished from the common appearance of halos seen around numerous cells in the normal reactive brain or other types of tumors where the nucleus is often not centrally located and the phenotype is seen only focally and not all throughout the tumor. Oligodendrogliomas are frequently associated with microcalcifications and the tumor cells can cluster around vessels and neurons in a visually striking “satellite” arrangement.
IHC, while not officially part of the current WHO diagnostic criteria for oligodendrogliomas, is extremely helpful in identifying these tumors. All oligodendrogiomas are positive for the oligodendroglial lineage marker OLIG2 as well as IDH1(R132H) (see molecular profile below) [18]. The tumors should lack expression of TP53 in most cases. Each of these markers typically has a homogeneous expression pattern with the marker being detected in essentially all of the tumor cells.
Grading of oligodendrogliomas follows WHO guidelines using cellularity, anaplasia, mitoses, vascular proliferation, and necrosis as the main criteria for assignment. Most oligodendroglioma WHO Grade II have few to no mitoses, no more than moderate cell density and generally very minimal atypia. Tumors are categorized as anaplastic oligodendroglioma WHO Grade III if brisk mitotic activity, necrosis, or vascular proliferation is present. The criteria for this are somewhat controversial and distinction from higher grade glioblastoma can sometimes be a challenge as not all of these features are generally present in high-grade tumors. Specific stipulations as to how many should be present are not yet established. No grading-specific markers have been identified for oligodendrogliomas but could be useful for future evaluation of these tumors.
16.5.3 Astrocytoma
Astrocytomas are diffuse gliomas that are common in children and young adults. They were originally named based on the tumor cell resemblance to non-tumorigenic reactive astrocytes in the brain. However, more recent studies have shown that astrocytomas invariably contain a mixture of both astrocytic and oligodendroglial lineage cell types with varying degrees of abundance of each. The WHO entity termed “Oligoastrocytoma” or mixed glioma, was created to further distinguish tumors which had more obvious oligodendroglial features [16]. However, oligoastrocytoma is now considered to lie within the spectrum of morphology seen in the category of astrocytomas, and in practice is not reliably able to be distinguished as a separate entity morphologically, biologically, or genetically. As a result, astrocytoma and oligoastrocytoma are expected to be merged into one category, defined by the presence of IDH1 mutations and absence of 1p/19q codeletion, in order to achieve a category with highly uniform genetics, biology, and clinical prognosis. These more rigorous requirements are the generally stated goal for all class distinctions in the next WHO consensus [19].
Overall, astrocytomas are easily recognized as distinct from oligodendrogliomas by their more pleomorphic and irregular nuclear cytology with mixtures of rounded and elongate cell shapes, the frequent presence of eosinophilic cytoplasm, and lack of significant perinuclear halos in tumor cells. The tumors are sometimes difficult to recognize in their lower grade forms and particularly in children [20]. The background neuropil is frequently intact due to their infiltrative nature although several variants and grades exist with varying alteration in the background.
Grading of astrocytomas is based on mitotic rate and the presence of vascular proliferation or necrosis. Diffuse astrocytomas WHO Grade II are the lowest form of this particular category and generally are slow growing with usually no mitoses identified in the tumor, a low cell density and pleomorphism. The presence of multiple mitoses designates a tumor in this class as WHO Grade III and is considered “high grade” for clinical treatment purposes. The presence of vascular proliferation or necrosis prior to treatment of the tumor promotes a tumor to WHO Grade IV and is then referred to as glioblastoma by current criteria.
16.5.4 Glioblastoma
The histology of the majority of glioblastomas (GBMs) is quite distinct from the lower grade astrocytomas (Grade II and Grade III) and this likely reflects their unique biology and general absence of mutations in IDH1/2 genes [21]. The typical case is de novo in origin with no prior precursor lesion in the patient (e.g., primary GBM). Secondary GBM can arise from a lower grade IDH1 mutant astrocytoma but it is estimated that fewer than 10 % of GBMs may arise from such progression. The histology of GBM is diverse. This was even indicated by their originally assigned name “glioblastoma multiforme.” Although the diversity is a key feature, they do in fact share several histological and clinical features which make them morphologically distinguishable from oligodendrogliomas and other CNS tumors.
By WHO 2007 classification, GBMs all fall under the larger category of diffuse astrocytomas and are the highest most aggressive Grade IV within this category. While originally thought to represent a “pure” astrocytic tumor and potentially even derived from astrocytes, recent studies have shown that all astrocytomas are somewhat misnamed. Like their lower grade astrocytoma counterparts, GBMs are all of a mixed astrocytic and oligodendroglial lineage as defined by multiple markers for these cell types (e.g., GFAP, OLIG2). They are likely to originate from several normally dividing populations of stem or progenitor cell types within the normal brain [18]. Like other diffuse gliomas, GBMs exhibit striking diffuse infiltration and intermingling with the preexisting brain structures making them incurable by surgical resection.
GBMs by definition have marked anaplasia, mitotic activity, necrosis, and microvascular proliferation which are the essential features used to distinguish GBM from lower grade astrocytomas (Fig. 16.3c and d). These features automatically allow grading as WHO Grade IV and this is the maximum grade allowed by the WHO. However, currently WHO guidelines recognize three histological subclasses: classic GBM, gliosarcoma, and giant cell GBM. Additional subclasses have been described in the literature and include GBM with oligodendroglioma component, small cell GBM, and epithelioid/adenoid GBM but have not yet been accepted as distinct or able to be reliably distinguished as clinico-pathologic or molecular entities. Although these subclasses are commonly referred to in practice, as yet none are associated with significant prognostic value.
16.6 Molecular Pathology of Brain Tumors
16.6.1 Diagnostic Tools
There has been an exciting explosion in the number and type of assays available to pathologists for improved molecular and cytogenetic analysis of cancer in the past decade and many of these are now routinely used in brain tumor diagnostics to provide prognostic and treatment information [22]. While all the methods were first pioneered in the research setting, the advancement to clinical use requires careful consideration of factors such as accuracy, cost, and turnaround time, which, in the clinical realm can often be quite different than that required in the research laboratory setting. Assays to determine copy number, single nucleotide variants, and epigenetic assessments are all commonly used in the clinical setting and described below.
16.6.1.1 RNA and Protein Expression
RNA is a labile biomarker which has seen rather limited adoption in the clinical realm. RNA in situ hybridization is utilized in a research setting but no brain tumor biomarkers of clinical relevance have been discovered. While RNA expression profiling has been useful in the research setting, it has not seen rapid adoption with modern techniques commonly used in the clinic and most clinical assessments are performed to detect protein expression via IHC methods (Fig. 16.4a and b) [23, 24].
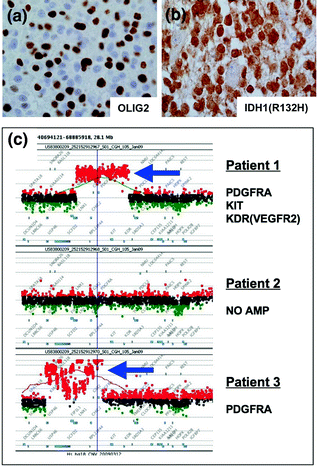
Fig. 16.4
Immunohistochemical and molecular methods for assessing brain tumors. a Immunohistochemistry for OLIG2 is positive (brown) in a subpopulation of astrocytoma cells. b IDH1(R132H) mutation (brown) in an astrocytoma demonstrates strong uniform expression by IHC, c array CGH results showing PDGFRA locus in patients with glioblastoma. Patient 1 shows amplification involving PDGFRA, KIT, and KDR, patient 2 shows no aberration, and patient 3 shows amplification involving only the PDGFRA gene with a complex pattern. Red probes = gains, green probes = losses, black = neutral; blue arrows indicate amplification events
16.6.1.2 Cytogenetics
Cytogenetic techniques were some of the first methods employed to clinically distinguish gains and losses of DNA and specific regions of the genome or genes that were altered. Karyotypes from acutely cultured cells were used to determine associated gains or losses of chromosomes (e.g., gain of Chr 7 and loss of Chr 10 in GBM) in several brain tumors, however the application of this in practice was fairly limited compared to other cancers due to its fairly limited clinical value and is no longer commonly used in the diagnostic setting.
Fluorescence in situ hybridization (FISH), introduced in Chap. 2, is an extremely useful technique for hybridization and counting of copies of specific probes on tissue sections at targeted regions of the genome. This analysis has the advantage of being analyzed at a single cell level and, therefore, is extremely sensitive and specific. It is one of the most commonly used methods today to assess brain tumor copy aberrations, but given its targeted nature of analysis of only single regions of the genome, it is being replaced by more multiplexed or whole genome approaches.
Hybridization based copy number arrays (i.e., array CGH, SNP arrays) are highly sophisticated tools for multiplex assessment of the number of copies of genes or loci in the genome. These technologies now allow pathologists to simultaneously query up to one million different locations with good sensitivity and specificity in clinical FFPE samples (Fig. 16.4c) [25]. Next generation sequencing panels are also now used to assess copy number with good results for large (arm level) copy changes in cancers but has more limited ability to reliably detect smaller size and low level changes in copy number [26]. As example, there is often poor sensitivity for small single copy gains and deletions due to the cost of reaching broad coverage as is typically achieved in array based assays. New analytical methods and lowered sequencing costs are expected to improve this situation in the future.
16.6.1.3 Sequencing
Detection of single nucleotide variants (e.g., point mutations) was originally performed using Sanger sequencing and other targeted methods and has been most commonly used to identify mutations in IDH genes [27]. Targeted sequencing panels based on PCR amplification of multiple genes of interest are also commonly used in current practice [28]. However, the advent of next generation multiplex sequencing methods has rapidly advanced the ability to detect multiple mutations in cancer simultaneously and has been rapidly adopted for diagnostics and prognostic use in brain tumors [26, 29].
Most sequencing is currently focused on specific known oncogenes and tumor suppressors either at “hotspots” or at all exonic sequences of these genes, in order to efficiently detect mutations of interest in FFPE samples. The use of the targeted exome sequencing approach is particularly useful for brain tumors where tumor suppressor genes are frequently altered and lack consistent “hotspots” for effective evaluation. Current focus on next generation sequencing methods are on reduced turnaround time to allow more rapid use in clinical practice attempting to balance increased gene coverage with time required for analysis. The next generation sequencing techniques also allow for assessment of gene insertions or deletions (indels) and gene fusion events, however, analytical tools for such events are currently not reliable or widely available for clinical practice. These are expected to evolve rapidly to enable such events to be detected with clinical standards of sensitivity and specificity in the near future.
16.6.1.4 Epigenetics
Brain cancers were one of the first indications where assessment of epigenetic modifications of the genome were of clinical relevance, specifically with respect to methylation status of the methylguanine methyltransferase (MGMT) gene in gliomas [30, 31]. Methylation events are characteristically assessed clinically using methylation specific PCR strategy with bisulfite conversion of methylated cytosine to uracil. The converted DNA is then sequenced using Sanger or pyrosequencing methods to infer which residues in the target regions of interest were methylated. As for copy number and single nucleotide variant detection methods, the use of multiplexed methylation arrays capable of whole genome methylation assessments has recently been entering into clinical use. Given the association of global methylation patterns with cell lineage, methylation arrays are most commonly used currently for recognition of diagnostic patterns of methylation in specific tumor types [32, 33]. Also such methods can reliably report out copy number information at the same time, in a similar manner to dedicated copy arrays. Interestingly, use of methylation arrays has not replaced targeted approaches for MGMT promoter assessment, although with further improvements and experience this is expected to be possible in the future.
16.6.2 Cytogenomic Features of Brain Tumor Types
16.6.2.1 Meningiomas
The most common molecular events in meningiomas are loss of the tumor suppressor gene NF2 on chromosome 22q, which occurs in approximately 50 % of sporadic and nearly 100 % of syndromic tumors. Atypical WHO Grade II meningiomas are associated with losses of 1p and 14q most commonly. The overall burden of copy number changes appears to increase with histologic grade and recurrence risk in atypical meningioma and following gross total resection could be predicted using array CGH or other multiplex copy number assays to determine the overall genomic copy number changes within the tumor [34].
Recent sequencing studies of meningioma have identified strong clinico-pathologic correlation of mutational genotypes with histologic subtypes of meningiomas likely due to cell of origin differences. Oncogenic mutations in AKT1 and SMO were typically associated with an anterior/medial skull base location [35]. Mutations in TRAF7, a pro-apoptotic E3 ubiquitin ligase, have also been described in approximately 25 % of meningiomas as a common recurrent event and were present in association with KLF4 mutations or less commonly AKT1 mutations [36]. Drugs targeting the NF2, SMO, and AKT1 pathways are currently being evaluated in clinical trials of meningiomas and may help to address the lack of effective medical therapies for this disease.
16.6.2.2 Oligodendroglioma
These tumors were one of the first brain tumors to be recognized as associated with a specific diagnostic and prognostic molecular genetic signature. Essentially, all oligodendrogliomas exhibit codeletion of chromosomes 1p and 19q which are present in greater than 90 % of cases in carefully selected series [21]. The codeletion of these chromosomal arms was noted to result from a balanced translocation with subsequent loss of one rearranged allele leading to overall single copy loss of these regions [37]. More recently, these tumors were also noted to universally harbor mutations in either the IDH1 or IDH2 genes [21, 38]. These mutations alter the enzymatic activity of the IDH proteins leading to high levels of the onco-metabolite 2-hydroxyglutarate (2HG) which is normally expressed at only low levels in cells and may promote tumor growth. Furthermore, IDH mutations are strongly associated with a distinct methylation profile termed G-CIMP [39]. Such a profile may be induced by the IDH mutations or may represent the profile of the cell of origin for these tumors that has yet to be defined.
Other mutations in CIC, an HMG-box transcription factor with repressor activity, are identified in approximately 70 % of oligodendrogliomas and are unique to the disease. The presence of mutations in ATRX, TP53, and amplification of EGFR, hallmarks of other gliomas, are extremely rare in oligodendroglioma and typically 1p/19q codeletion are mutually exclusive with each of these events. While not yet formally utilized in the grading of oligodendrogliomas, the presence of CDKN2A loss is the most prominent change identified.
16.6.2.3 Astrocytoma
Astrocytomas are first defined by the presence of IDH1 mutations, most commonly the IDH1(R132H) variant (more than 80 % of patients). At a very high rate IDH1 mutations in this disease co-occur with loss of function mutations in the tumor suppressor genes ATRX and TP53 [21]. The most common mutations in ATRX lead to loss of expression of the protein in tumor cells as detected by IHC staining for the wild-type protein and is a useful and cost effective assay in clinical practice. Conversely, the most common mutations in TP53 lead to increased expression of P53 in tumor cells by IHC, with more than 10 % of cells generally having high-level nuclear expression. However, the use of these IHC markers as surrogates for detection of IDH1/2, ATRX, or TP53 mutations in oligodendrogliomas or astrocytomas can miss approximately 10–20 % of those cases with variant mutations whose nature does not alter protein expression in the predicted manner. Multiplex sequencing methods are, therefore, highly useful for reliable assignment of prognosis and diagnosis in all gliomas.
16.6.2.4 Glioblastoma
GBM is one of the best characterized cancers at the molecular level [40] and was one of the first cancers to be studied in large scale integrative genomic approaches of The Cancer Genome Atlas (TCGA) program established by the National Cancer Institute [41]. Collectively, these molecular studies have consistently identified three core signaling pathways which are disrupted in GBM: increased activation of receptor tyrosine kinase/RAS/PI3 K signaling, loss of function in P53 signaling, and reduced signaling of the RB pathway. Aberrant activation of RTK/RAS/PI3 K signaling is evident in 88 % of GBMs and most characteristically occurs due to amplification of the EGFR gene, along with rearrangements and overexpression of mutant EGFRvIII and extracellular domain mutants [42]. This pathway is also activated by amplification of other well known oncogenes including PDGRA, MET, AKT, or PIK3CA and/or aberrations that lead to loss of the PTEN tumor suppressor gene function. Alterations in the tumor suppressor TP53 are common in a subset of adult GBM including at least 42 % of adult tumors, either through direct mutations in TP53 or by amplification of MDM2 or MDM4 each seen in approximately 10 % of patients. Interestingly, with time studies have shown that TP53 mutations are more common in young adult astrocytomas and pediatric high-grade gliomas where they are seen in the majority of these tumors [43, 44]. The RB pathway is frequently targeted by different routes, including genomic losses combined with mutation of RB, as well as genomic losses of the CDKN2 family of tumor suppressor genes, or amplification of the negative regulators of the RB pathway, such as CDK4.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


