Chapter 14 Pathobiology Clinical Features, And Management Of Sickle Cell Disease
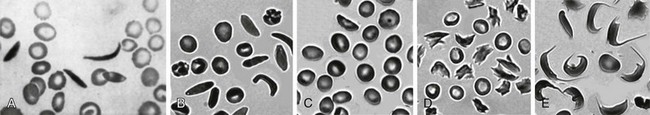
Figure 14-1 SICKLE RED BLOOD CELL (RBC) MORPHOLOGIES.
(B to E from Obata K, Mattiello J, Asakura K, et al: Exposure of blood from patients with sickle cell disease to air changes the morphological, oxygen-binding, and sickling properties of sickled erythrocytes. Am J Hematol 81:26, 2006. A from Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch Intern Med 5:517, 1910.)
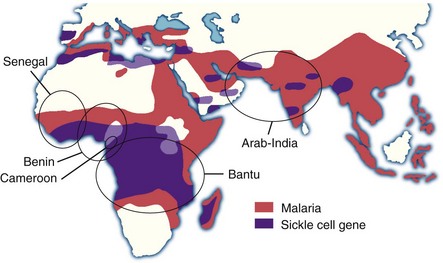
Figure 14-2 SICKLE GENE AND MALARIA.
(Adapted from Friedman MJ, Trager W: The biochemistry of resistance to malaria. Sci Am 244:154, 1981 and from Nagel RL, Steinberg MH: Genetics of the βS gene: Origins, epidemiology, and epistasis in sickle cell anemia. In Steinberg MH: Forget BG, Higgs DR, Nagel RL, eds: Disorders of hemoglobin: Genetics, pathophysiology, and clinical management, Cambridge, 2001, Cambridge University Press, p 711.)
Relationship of HbS Molecular Behaviors to Disease Features
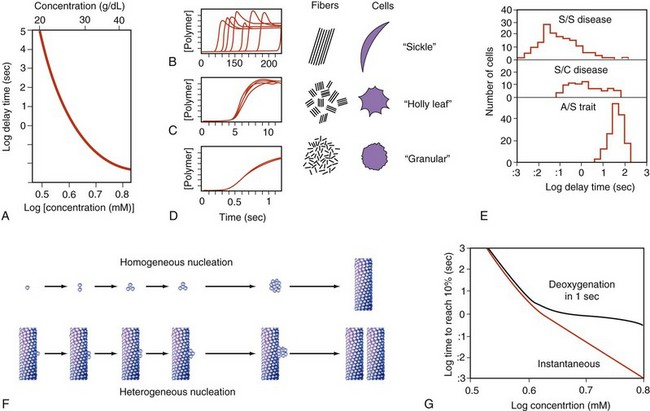
Figure 14-3 KINETICS OF HEMOGLOBIN S POLYMERIZATION, STUDIED BY NEAR-INSTANTANEOUS AND COMPLETE DEOXYGENATION.
A, Extreme dependence of delay time on hemoglobin concentration. B to D, Kinetic progress curves for polymer formation show that long delay times are highly variable (B), but very short delay times are highly reproducible (D). To the right is a representation of domains and corresponding red blood cell morphology postulated to result from these different scales of polymerization rate (see Fig. 14-1, B to E). E, Delay times for individual red blood cells are influenced by substituent hemoglobins. F, A double nucleation process is hypothesized to underlie polymer formation. G, Physiologically, the finite rate of deoxygenation effectively caps the polymerization rate and eliminates the relevance of delay times that are short relative to deoxygenation rate (<1 sec).
(A to E, Data from Eaton WA, Hofrichter J: Hemoglobin S gelation and sickle cell disease. Blood 70:1245, 1987; F adapted from Ferrone FA, Hofrichter J, Eaton WA: Kinetics of sickle hemoglobin polymerization II. A double nucleation mechanism. J Mol Biol 183:611, 1985; G, Data from Ferrone FA: Oxygen transits and transports. In Embury S, Hebbel RP, Mohandas N, Steinberg MH, eds: Sickle cell disease: basic principles and clinical practice, New York, 1994, Raven Press.)
Major Sickle RBC Membrane Defects
Enhanced mechanosensitivity → ↑ deformability responses →
Abnormal RBC adhesion to endothelium
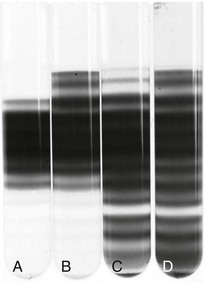
Figure 14-4 MARKED HETEROGENEITY IN SICKLE RED BLOOD CELL (RBC) HYDRATION.
(From Embury SH, Clark MR, Monroy G, Mohandas N: Concurrent sickle cell anemia and a-thalassemia. J Clin Invest 73:116, 1984.)
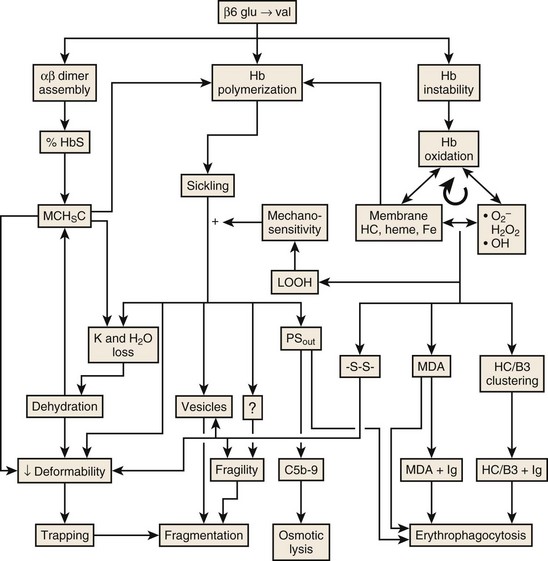
Figure 14-5 MECHANISMS LEADING TO HEMOLYSIS IN SICKLE CELL DISEASE.
(Modified with permission from The American Journal of Hematology from Hebbel RP: Reconstructing sickle cell disease: A data-based analysis of the “hyperhemolysis paradigm” for pulmonary hypertension from the perspective of evidence-based medicine. Am J Hematol
Related posts:
 Dynamic Interactions between Hematopoietic Stem and Progenitor Cells and the Bone Marrow: Current Biology of Stem Cell Homing and Mobilization
Clinical Manifestations and Treatment of Acute Lymphoblastic Leukemia in Children
Approach to Anemia in the Adult and Child
Infectious Mononucleosis and Other Epstein-Barr Virus–Associated Diseases: Part 2
Hematologic Changes in Pregnancy
Pain Management and Antiemetic Therapy in Hematologic Disorders
Dynamic Interactions between Hematopoietic Stem and Progenitor Cells and the Bone Marrow: Current Biology of Stem Cell Homing and Mobilization
Clinical Manifestations and Treatment of Acute Lymphoblastic Leukemia in Children
Approach to Anemia in the Adult and Child
Infectious Mononucleosis and Other Epstein-Barr Virus–Associated Diseases: Part 2
Hematologic Changes in Pregnancy
Pain Management and Antiemetic Therapy in Hematologic Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


