9.1
Introduction
Parathyroid hormone (PTH) and PTH–related protein (PTHrP) are major polypeptide factors that regulate skeletal physiology and mineral homeostasis. The appearance of the parathyroid glands during the evolution of terrestrial vertebrates underscores the primary functional role of PTH—the maintenance of adequate levels of plasma-ionized calcium in the face of a calcium-deficient terrestrial environment. The secretion of PTH by the parathyroid glands is stimulated when plasma-ionized calcium levels fall. Once secreted, PTH acts to restore normal levels of ionized calcium through an integrated series of actions on bone, kidney, and (indirectly) the intestine. PTHrP, when present as a circulating factor, produces target cell effects that resemble those of PTH. This is most evident in malignancy-associated hypercalcemia where tumors elaborate sufficient quantities of PTHrP to produce biochemical abnormalities overlapping those seen in primary hyperparathyroidism. However, the major physiologic function of PTHrP is to act as a local (paracrine) factor that controls the development, morphogenesis, and function of a variety of tissues including (but not limited to) those involved in skeletal and mineral homeostasis. PTH and PTHrP are tied together historically in which PTHrP was discovered as a result of the quest to understand the pathogenesis of malignancy-associated hypercalcemia. However, they are also related structurally and produce their major physiological effects by activating a common receptor, the PTH/PTHrP receptor. The present chapter focuses on current understanding of the physiology and mechanism of action of these two polypeptides. For excellent reviews on PTH and PTHrP, see Refs. .
9.2
Synthesis and secretion of parathyroid hormone
The gene encoding PTH in humans is on the short arm of chromosome 11. The translation product of the PTH gene, preproPTH, is converted to proPTH and finally to the mature 84 amino acid, biologically active secretory product PTH . The parathyroid glands store limited amounts of PTH, but de novo synthesis is required to maintain chronic PTH secretion. During early development the parathyroid glands derive from the third and fourth endodermal pharyngeal pouches in response to signals from the sonic hedgehog and fibroblast growth factor (FGF) pathways. These signals lead to the expression of the master transcription factor glial cells missing-2 (GCM2) which drives the differentiation and survival of parathyroid cells . The parathyroid glands first appear during evolution with the movement of animals from an aquatic to a terrestrial environment deficient in calcium. Maintenance of adequate levels of plasma-ionized calcium (1.0–1.3 mM) is required for normal neuromuscular function, bone mineralization, and many other physiologic processes. The parathyroid gland secretes PTH in response to very small decrements in blood ionized calcium in order to maintain the normocalcemic state. As discussed later, PTH accomplishes this task by promoting bone resorption and releasing calcium from the skeletal reservoir; by inducing renal conservation of calcium and excretion of phosphate; and by indirectly enhancing intestinal calcium absorption by increasing the renal production of the active vitamin D metabolite 1,25(OH) 2 vitamin D. The parathyroid gland functions in essence as a “calciostat,” sensing the prevailing blood ionized calcium level and adjusting the secretion of PTH accordingly ( Fig. 9.1 ) . The relationship between ionized calcium and PTH secretion is a steep sigmoidal one, allowing significant changes in PTH secretion in response to very small changes in plasma-ionized calcium.
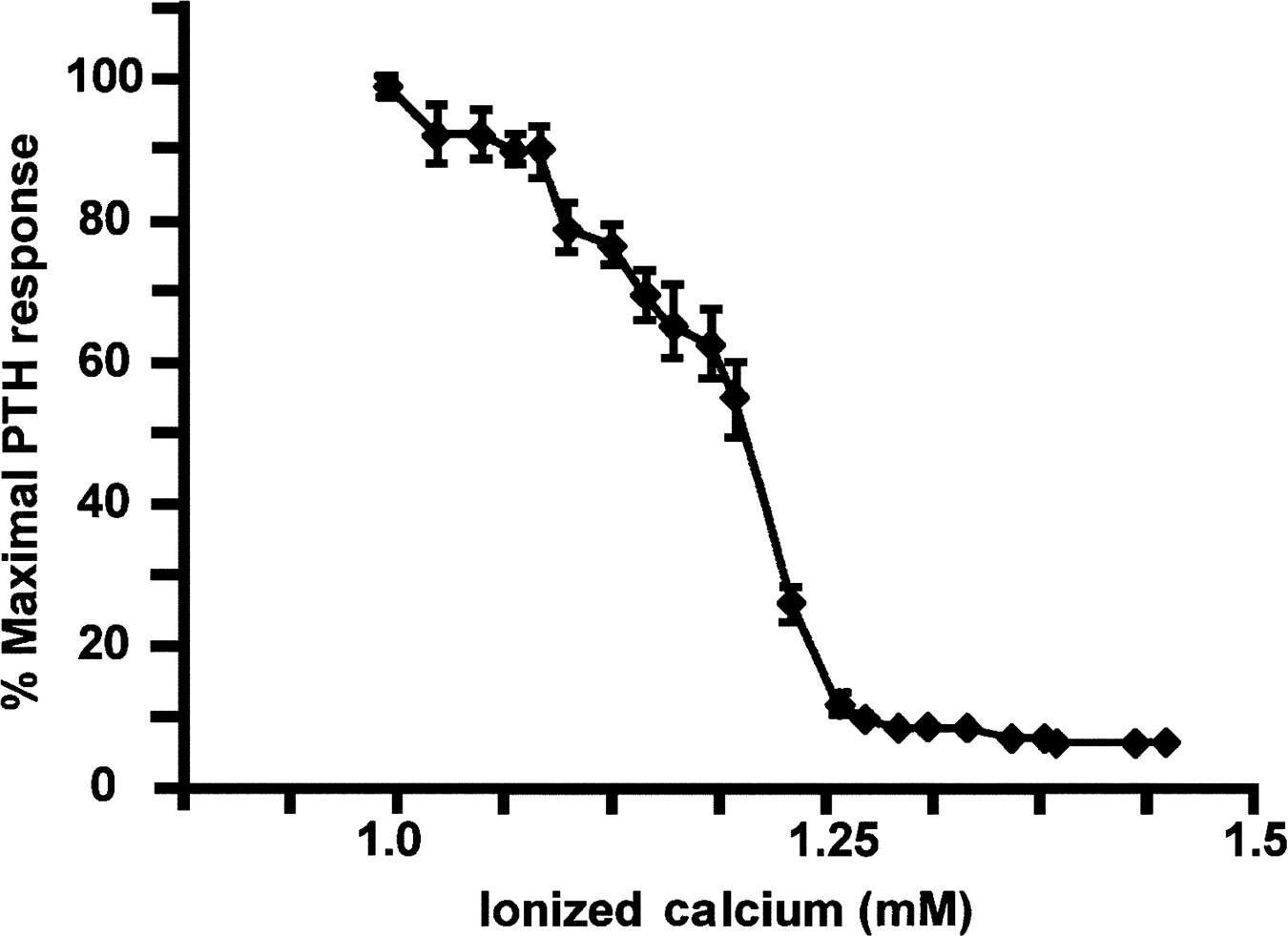
The prevailing level of serum ionized calcium regulates both the acute and chronic secretion of PTH. Sustained hypocalcemia promotes increased expression of the PTH gene associated with stabilization of PTH messenger ribonucleic acid (mRNA) as well as increased parathyroid cell proliferation . The latter accounts for the marked parathyroid hyperplasia (secondary hyperparathyroidism) that frequently accompanies chronic renal failure. 1,25(OH) 2 vitamin D also serves as a negative regulator of PTH gene expression and parathyroid cell hyperplasia. In chronic renal failure, both hypocalcemia and reduced circulating levels of 1,25(OH) 2 vitamin D presumably contribute to the progression of secondary hyperparathyroidism . It is noteworthy that 25(OH) vitamin D can suppress PTH gene transcription , possibly through the local production of 1,25(OH) 2 vitamin D by 25(OH) vitamin D-1-hydroxylase present in the parathyroid gland .
The mechanism by which parathyroid cells respond to changes in plasma calcium is through a plasma membrane calcium-sensing receptor (CaSR). Unlike intracellular calcium-binding proteins, which have an affinity for free calcium in the nanomolar range (consistent with intracellular levels of free calcium), the CaSR binds calcium in the millimolar range. This receptor, first identified in 1993 , is a member of the class C G protein-coupled receptor (GPCR) superfamily and contains calcium-binding elements in its extracellular domain, and signaling determinants in its cytoplasmic regions . Calcium binding to the receptor triggers activation of the G-proteins Gq/G 11 and (to a lesser extent) Gi, resulting in stimulation of phospholipase C (PLC) and inhibition of adenylyl cyclase, respectively. This results in an increase in intracellular calcium and a decrease in cyclic adenosine monophosphate (cAMP) levels in parathyroid cells. By mechanisms that are not yet clear, these signaling pathways serve to suppress the synthesis and secretion of PTH. When blood ionized calcium falls, there is less signaling by the CaSRs on the parathyroid cell and PTH secretion consequently increases. The essential role of the CaSR can be clearly seen in humans bearing loss-of-function mutations in the CaSR gene . In the heterozygous state, such mutations result in familial hypocalciuric hypercalcemia, characterized by inappropriately high levels of PTH secretion in the face of mild hypercalcemia. These individuals are quantitatively resistant to the suppressive effect of calcium on PTH secretion due to the reduced number of parathyroid CaSRs. In the homozygous state, patients display a large increase in PTH secretion with life-threatening hypercalcemia (neonatal severe primary hyperparathyroidism). Mice with homozygous and heterozygous disruption of the CaSR gene display phenotypes similar to those seen in humans . Point mutations in the CaSR or in the alpha subunit of G 11 that produce constitutive signaling have also been described, and these are associated with autosomal-dominant hypocalcemia in humans .
Pharmacological ligands for the CaSR have been developed, and these are effective in altering the ability of the CaSR to signal . Calcimimetic drugs bind to transmembrane regions in the CaSR and increase the receptor’s sensitivity to extracellular calcium. This results in an increase in receptor signaling and thus suppression of PTH secretion. Calcimimetic drugs have clinical utility in the medical management of hyperparathyroidism . Calcilytic drugs act as pharmacological antagonists of the CaSR, thereby increasing the secretion of PTH. Short-acting calcilytic drugs are capable of stimulating bone formation in rodents presumably due to transient increases in PTH secretion . However, it remains to be seen whether this is a viable approach for treating osteopenia in humans.
Hyperphosphatemia, as occurs in the late stages of chronic kidney disease, is associated with hyperparathyroidism with parathyroid gland hyperplasia. Phosphate forms a complex with ionized calcium in the circulation, thus reducing the level of ionized calcium, and this results in increased synthesis and secretion of PTH as well as increased proliferation of parathyroid cells . High serum phosphate also appears directly to promote PTH secretion by stabilizing PTH mRNA . Recently, a binding site for phosphate in the CaSR has been described, and phosphate binding to this site appears to stabilize the inactive conformation of the receptor, thus promoting PTH secretion . Another potential regulator of PTH secretion is FGF23, an endocrine FGF with phosphaturic activity that is secreted by osteoblast-lineage cells in response to increased dietary phosphate intake. FGF23 has been shown to act on the parathyroid gland to inhibit the secretion of PTH, providing a possible physiological link between phosphate homeostasis, osteoblast function, and PTH secretion .
9.3
Metabolism of parathyroid hormone
The metabolism of PTH has been the subject of multiple review articles . It has been known for decades that PTH circulates in multiple forms that can be distinguished by radioimmunoassays specific for different regions of the PTH molecule . This heterogeneity has two origins ( Fig. 9.2 ). PTH(1–84) is subject to metabolism within the parathyroid gland, resulting in secretion of PTH fragments as well as the intact molecule. In addition, PTH(1–84) is metabolized in peripheral tissues. Mid-region and C-terminal fragments of PTH have a much longer half-life in the circulation than does PTH(1–84) . As a result, mid-region and C-terminal fragments of PTH circulate at much higher concentrations than intact PTH(1–84) . Rapid plasma clearance of PTH is due primarily to hepatic metabolism, with a lesser contribution by the kidneys . Peripheral metabolism generates mid- and C-terminal fragments of PTH that resemble those secreted by the parathyroid gland. Mid- and C-terminal PTH fragments are cleared by renal excretion, and thus circulating the levels of these fragments are highly dependent upon renal function. Extremely high levels of PTH detected with antibodies against the mid- and C-regions of the hormone in many patients with end-stage renal disease thus reflects a combination of secondary hyperparathyroidism and reduce renal clearance of PTH fragments.
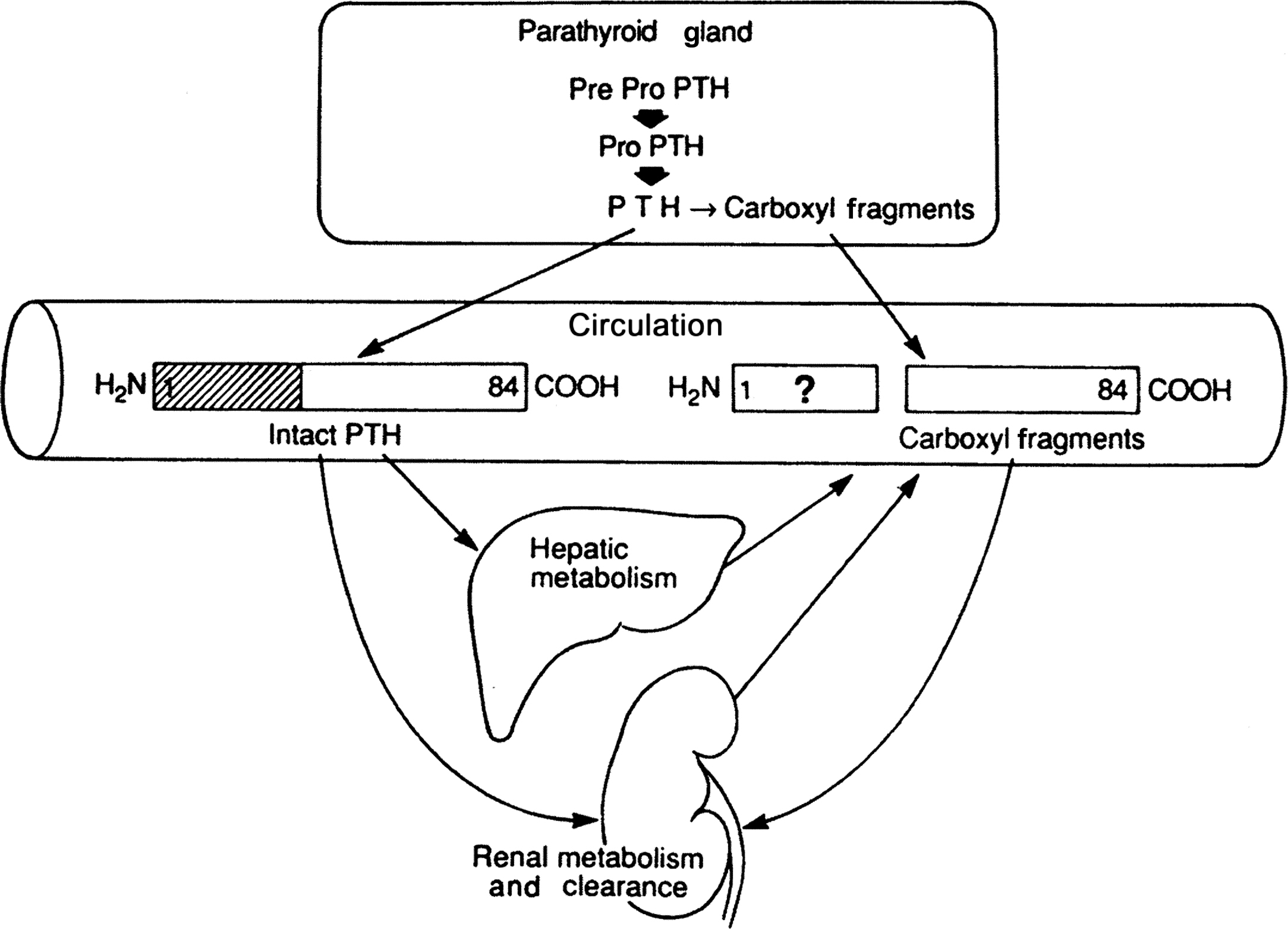
Mid- and carboxyl-region PTH fragments lack the N-terminal 1–34 sequence of the hormone required for binding to PTH/PTHrP receptors and producing the classical effects of PTH on kidney and bone. Metabolism of PTH could produce biologically active, N-terminal fragments of PTH, but there is little evidence for the presence of significant levels of N-terminal PTH fragments in the circulation , or for significant secretion of such fragments by the parathyroid gland . Presumably, both the parathyroid gland and peripheral organs contain enzymes that degrade N-terminal fragments of PTH. This ensures that the circulating levels of biologically active PTH are derived exclusively from glandular secretion of PTH(1–84). There is some limited evidence for cellular effects of mid- or C-region fragments of PTH, but their biological role if any remains unclear.
Calcium-sensitive cathepsins are responsible for cleaving PTH(1–84) within the parathyroid gland. Intraglandular cleavage occurs between residues 34 and 35 or between residues 36 and 37 , and a greater proportion of PTH is cleaved under conditions of hypercalcemia . The N-terminal fragments so produced are rapidly degraded within the parathyroid gland, and thus calcium-sensitive cleavage constitutes a mechanism for inactivation of PTH. Therefore the level of plasma calcium determines not only the rate of synthesis and secretion of PTH but also the extent to which secreted PTH is biologically active.
A large fragment of PTH identified as PTH(7–84) has been identified in the circulation . This fragment is secreted from the parathyroid glands following calcium-dependent intraglandular proteolysis of the N-terminus of PTH(1–84) . It may also arise from peripheral metabolism of PTH(1–84) . PTH(7–84) lacks the N-terminal residues required for activation of PTH/PTHrP receptors. However, this fragment is detected in some radioimmunoassays for “intact” PTH resulting in overestimation of levels of circulating, biologically active PTH . PTH(7–84) is known to bind with low affinity to PTH/PTHrP receptors, thereby antagonizing the actions of PTH(1–84) . However, it remains uncertain whether endogenous PTH(7–84) circulates at levels sufficient to suppress the target cell actions of PTH under normal physiological conditions.
9.4
Bone-resorbing action of parathyroid hormone
PTH serves to maintain adequate plasma calcium in the face of a calcium-deficient terrestrial environment. When dietary calcium intake is inadequate, PTH maintains the level of plasma calcium by mobilizing calcium from the vast reservoir present in bone in the form of hydroxyapatite mineral. PTH appears to accomplish this by promoting bone resorption that mobilizes calcium from bone mineral into a soluble form and by increasing the efflux of soluble calcium from bone to blood. Administration of PTH produces rapid movement of calcium out of bone, an effect that is associated with structural changes in cells lining the endosteal surface. It has been suggested that these lining cells form an epithelial-like barrier between the circulation and the bone extracellular fluid , and PTH may act on these cells to promote calcium transport. PTH enhances osteoclastic bone resorption within 15 minutes of its administration and produces a sustained increase in bone resorption that requires the differentiation of new osteoclasts. PTH-induced bone resorption involves the dissolution of hydroxyapatite bone mineral in the acidic microenvironment created by the osteoclast, as well as the degradation of collagen and other matrix proteins by proteolytic enzymes.
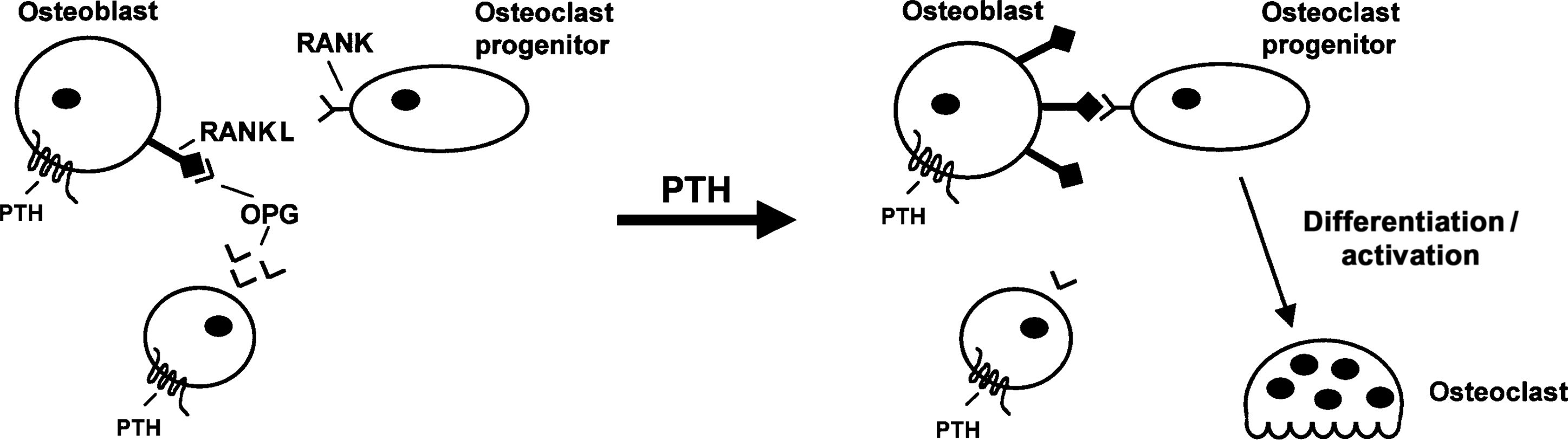
The action of PTH to promote bone resorption is crucial to the maintenance of systemic calcium homeostasis. PTH exerts this effect by promoting the differentiation and functional activity of osteoclasts, an action that occurs indirectly through the effects of PTH on osteoblast-lineage cells. The major mediator of this effect of PTH is receptor activator of nuclear factor kappaB ligand (RANKL). RANKL is up-regulated in response to PTH and acts on a tumor necrosis factor (TNF)-α receptor-related protein [receptor activator of nuclear factor kappaB (RANK)], which is expressed in myeloid precursors of osteoclasts as well as in differentiated osteoclasts . RANK signaling in osteoclast precursors promotes their differentiation to functional osteoclasts, and RANK signaling in differentiated osteoclasts enhances bone resorption and inhibits apoptosis . RANKL is required for normal osteoclast development and function, and mice lacking RANKL show a loss of functional osteoclasts resulting in osteopetrosis . Cells in the microenvironment of bone also secrete a truncated TNF-α receptor-like molecule termed osteoprotegerin (OPG) that functions as a “decoy receptor” by binding to RANKL and thereby preventing initiation of RANK signaling . The importance of OPG as a tonic suppressor of bone turnover is evident from findings with mice lacking functional expression of OPG. These animals display increased bone resorption and osteoporosis .
It is widely accepted that the RANKL/OPG/RANK system plays a preeminent role in PTH-induced bone resorption and calcium mobilization ( Fig. 9.3 ). Administration of soluble RANKL to mice elicits severe hypercalcemia within 1 day of administration, and increased osteoclast activity and bone loss are evident within 3 days . Administration of OPG (RANKL antagonist) blocks the calcemic action of exogenous PTH in vivo . Addition of OPG also inhibits PTH-induced osteoclast activation and bone resorption in vitro and in vivo . PTH produces an increase in the ratio of RANKL:OPG expressed by osteoblastic cells, an effect that is due to the ability of PTH to increase the expression of RANKL and to inhibit the expression of OPG . Similar effects are observed in vivo following the exogenous administration of PTH . The effect of PTH on RANKL is exerted at the level of gene transcription . However, this action of PTH is very rapid (evident within 1 hour) and thus up-regulation of RANKL could contribute not only to osteoclastogenesis but also to the rapid increase in the activity of mature osteoclasts seen in response to PTH. RANKL is produced as a membrane-associated protein, but it can be cleaved by proteases to yield a soluble form. The relative biological importance of these two forms under physiological circumstances is unclear. However, it has recently been shown that matrix metalloproteinase MMP14 (which can generate soluble RANKL) is up-regulated by PTH and that this is required for maximal PTH-stimulated bone resorption .
Other osteoclastogenic factors such as TNF-α and monocyte chemoattractant protein-1 (MCP-1) are up-regulated by PTH , but these are likely to play a minor role in mediating the control of bone resorption by PTH under physiological conditions. Recent studies have suggested that MCP-1 contributes significantly to bone resorption in a mouse model of hyperparathyroidism . It has been demonstrated that PTH action in Th17 T cells is required for the full catabolic response to PTH . It appears that PTH can promote the production of IL-17 by Th17 T cells, and this in turn enhances PTH-induced production of RANKL by osteoblast-lineage cells . Further studies are needed to define the role of T cells in the bone-resorbing action of PTH in humans.
There has been considerable interest in identifying the precise nature of the osteoblast-lineage cell that is the primary target of the osteoclastogenic effect of PTH. Studies in adult mice have demonstrated that PTH action on its receptors in osteocytes is sufficient for PTH-induced bone resorption . It is likely that a similar mechanism will obtain in humans, but direct evidence on this point is currently lacking.
9.5
Effects of parathyroid hormone on bone formation
Administration of PTH receptor agonists intermittently to animals or humans produces a marked anabolic response of the skeleton . This represents the basis for the efficacy of the PTH receptor ligands, teriparatide and abaloparatide, for the treatment of postmenopausal osteoporosis . PTH promotes bone formation in both trabecular and cortical bone, and these actions are associated with increased trabecular thickness and increased bone strength . High concentrations of PTH are known to produce an increase in the number of osteoblasts, which results in part from the coupling between increased osteoclastic resorption and new bone formation. However, intermittent treatment with lower doses of PTH produces a positive effect on osteoblastic bone formation.
The cellular basis for the anabolic action of PTH in bone is complex and not fully understood. A major reason for this is that PTH appears to act at multiple sites all of which contribute to the anabolic effect ( Fig. 9.4 ). Osteocytes are clearly a crucial target for PTH-induced anabolism. Osteocytes express high levels of PTH receptors, and genetic deletion of these receptors from osteocytes in mice blunts the anabolic response to PTH . Conversely, expression of constitutively active PTH receptors in osteocytes results in mice with bones with increased trabecular and cortical bone, and increased bone formation rates . Osteocytes appear to control bone formation through the regulation of wnt signaling in the bone marrow compartment. Osteocytes produce wnt proteins such has wnt1 which has been implicated in the control of bone formation in mice and humans . PTH has been shown to up-regulate the gene for wnt4 which is known to have a proanabolic action . Osteocytes also secrete potent inhibitors of wnt signaling. The most notable of these are dkk1 and sclerostin, both of which inhibit canonical wnt signaling (a pathway that serves to stabilize beta-catenin, allowing activation of target genes required for bone formation). PTH potently downregulates both dkk1 and sclerostin , resulting in derepression of canonical wnt signaling. This in turn induces the expression of runx2 , the master transcription factor for osteoblast differentiation, thus promoting bone formation.
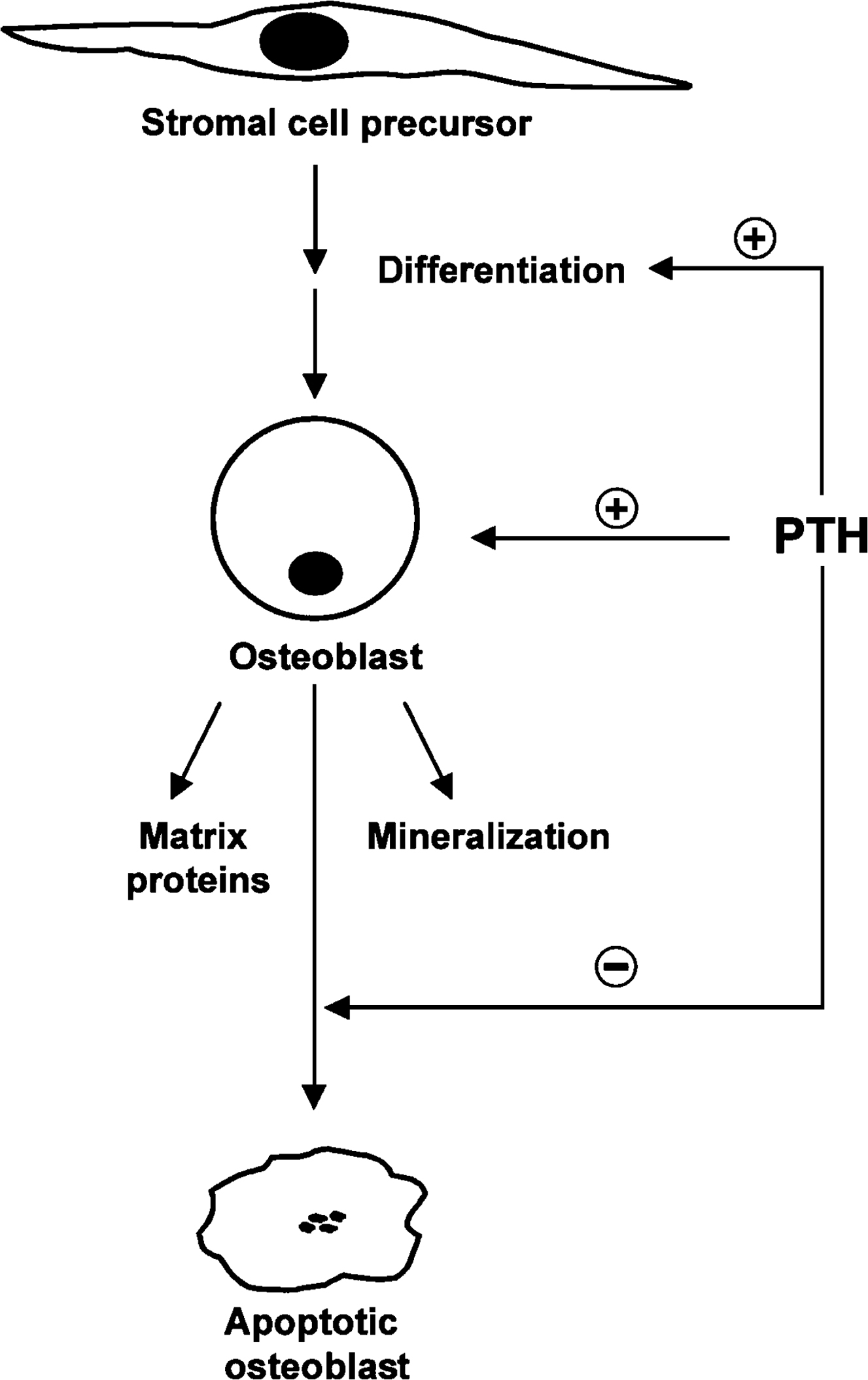
Nonosteocytic targets of PTH have also been implicated in the anabolic response. Mesenchymal progenitor cells express receptors for PTH, and PTH receptor signaling in these cells favors an osteogenic rather than an adipogenic fate possibly due to the activation of canonical wnt signaling . Deletion of Gs-alpha (required for PTH receptor signaling) in differentiating osteoblasts appears to accelerate the osteoblast-to-osteocyte transition resulting in less bone formation by mature osteoblasts . Thus PTH may serve to lengthen the duration over which osteoblasts are fully functional with respect to bone formation. In a similar vein, PTH has been shown to inhibit osteoblast apoptosis . PTH has also been shown to act on bone lining cells. These cells are not active in forming new bone, but they can be converted to functional osteoblasts in response to PTH . Finally, PTH can act on T cells to promote the production of wnt10b and thereby activate canonical wnt signaling and promote osteogenesis .
9.6
Renal actions of parathyroid hormone
PTH produces multiple renal responses that synergize with its bone-resorbing action to maintain adequate levels of plasma-ionized calcium. The renal actions of PTH include the inhibition of renal phosphate reabsorption, stimulation of renal calcium reabsorption, and increased production of 1,25(OH) 2 vitamin D. Patients with primary hyperparathyroidism display hypophosphatemia and decreased renal tubular reabsorption of phosphate, whereas hypoparathyroid patients are hyperphosphatemic and have increased phosphate reabsorption. Phosphate forms a complex with free calcium in blood. Thus for a given level of serum calcium, ionized calcium will be reduced as serum phosphate increases. Under conditions of relative hypocalcemia (e.g., during chronic dietary calcium deficiency), PTH secretion is increased resulting in increased bone resorption. Both calcium and phosphate are released from hydroxyapatite during the process of bone resorption. By promoting renal excretion of phosphate, PTH facilitates a rise in ionized as well as total plasma calcium.
Phosphate reabsorption in the proximal renal tubule is dependent in part on the activity of the type IIa sodium phosphate cotransporter (NaPi-IIa) . The phosphaturic action of PTH derives from the action of the hormone to inhibit the function of this transporter . NaPi-IIa is located in the apical plasma membrane and permits the coupled transport of sodium and phosphate from the tubular lumen into the renal cell. Exposure of proximal tubular cells to PTH results in a reduced V max of the transporter, and this is associated with a decrease in the amount of the transporter in the apical plasma membrane . Acute exposure of the proximal tubular cells to PTH enhances the endocytosis and subsequent lysosomal degradation of NaPi-IIa, and this may be the major mechanism responsible for rapid PTH-induced inhibition of renal phosphate reabsorption . The stability of NaPi-IIa in the apical membrane is maintained by its interaction with a Na/H exchange-regulatory molecule NHERF-1 . PTH promotes the dissociation of NHERF-1 leading to rapid endocytosis of NaPi-IIa and consequent reduced phosphate reabsorption.
A second renal action of PTH is to increase renal calcium reabsorption, thus ensuring that only small amounts of calcium released during PTH-induced bone resorption are lost via urinary excretion . The major sites for this effect of PTH are in the distal convoluted tubule, the collecting tubule, and the thick ascending limb of Henle’s loop . Available evidence indicates that distal renal tubular calcium reabsorption is an active process that requires calcium influx through TRPV5 calcium channels located in the apical plasma membrane . Drugs that inhibit these channels are effective in blocking PTH-induced renal calcium reabsorption. Mechanistically, it appears that TRPV5 channels are inhibited via association with the calcium-binding protein calmodulin . By inducing the phosphorylation of TRPV5, PTH reduces the channel’s affinity for calmodulin and this increases calcium channel activity. In addition, PTH appears to increase the abundance of TRPV5 on the apical membrane by inhibiting its endocytosis . Calcium entering the distal renal tubular cell in this manner is transported across the cell bound to calbindin-D28K and is then transported into the extracellular compartment via a sodium–calcium exchanger present on the basolateral plasma membrane . PTH up-regulates the expression of both of these calcium transport proteins .
A third renal action of PTH is to indirectly promote intestinal calcium absorption by increasing circulating concentrations of 1,25(OH) 2 vitamin D. This vitamin D metabolite acts directly on intestinal epithelial cells to increase the efficiency of calcium (and phosphate) absorption. Primary hyperparathyroidism is commonly associated with increased circulating levels of 1,25(OH) 2 vitamin D, whereas reduced levels of this metabolite are present in hypoparathyroidism . PTH produces this effect by increasing the production of 1,25(OH) 2 vitamin D and by reducing its catabolism. The production of 1,25(OH) 2 vitamin D results from the activity of the 25(OH) vitamin D-1-hydroxylase enzyme located in the proximal renal tubule . The expression of the gene encoding this enzyme, Cyp27b1, is up-regulated by PTH by a transcriptional mechanism . Conversely, the activity of vitamin D-24-hydroxylase, an enzyme that catabolizes 1,25(OH) 2 vitamin D, is downregulated by PTH . The net result is that PTH increases the circulating levels of 1,25(OH) 2 vitamin D and thereby promotes intestinal calcium absorption.
9.7
Parathyroid hormone–related protein as a mediator of humoral hypercalcemia of malignancy
The frequent occurrence of hypercalcemia in individuals with a variety of malignancies has been recognized for many years. An important clue as to the pathogenesis of humoral hypercalcemia of malignancy (HHM) came with the recognition that many such individuals display increased excretion of renal-derived (nephrogenous) cAMP . Activation of the renal PTH receptor by elevated circulating levels of PTH in hyperparathyroidism was the only known cause of increased nephrogenous cAMP, and thus it was suggested that malignant tumors produce a factor that activates PTH receptors. Plasma concentrations of immunoreactive PTH were found to be low in patients with HHM, indicating that the relevant circulating factor was not PTH itself.
Using the activation of PTH receptors as an assay, multiple groups succeeded in isolating and ultimately identifying the PTH-like etiologic factor in HHM . This factor was termed PTHrP because of its ability to bind to and activate the PTH receptor, and because of its limited sequence similarity to PTH . The PTHrP gene is subject to alternative splicing, resulting in the production of three protein products ranging from 139 to 173 amino acids differing only in their C-terminal sequence . PTHrP is capable of reproducing the major target cell actions of PTH and (like PTH) does so via the N-terminal 34 amino acids or so of the protein. A comparison of the 1–34 sequences of PTH and PTHrP reveal significant amino acid homology, with identity in eight of the 13 N-terminal residues. Two of the known contact sites between PTH and the PTH/PTHrP receptor are within this 13 amino-acid homologous region , indicating that these ligands use very similar mechanisms to activate their common receptor. The molecular mechanisms underlying the overexpression of PTHrP by malignant tumors are poorly defined. As the mass of PTHrP-expressing tumor cells expands, systemic levels of PTHrP eventually increase sufficiently to allow the peptide to elicit endocrine effects on PTH/PTHrP receptors in bone and kidney, resulting in HHM. It is possible but not established that PTHrP may play an autocrine role in promoting tumorigenesis. There is stronger evidence, however, that PTHrP plays a significant role in promoting bone metastases by stimulating tumor cell migration and enhancing the metastatic environment in bone through increased bone resorption and facilitating production/release of tumorigenic growth factors .
9.8
Physiological roles of parathyroid hormone–related protein
Although PTHrP produces PTH-like target cell effects in patients with HHM, circulating levels of PTHrP are very low to undetectable in normal individuals. This, coupled with the widespread expression of the PTHrP gene in normal tissues, suggested that PTHrP was likely to have physiological functions as a local, paracrine factor rather than as a systemic hormone. Subsequent studies have confirmed that PTHrP indeed plays an important physiological role in a wide variety of tissues ( Table 9.1 ) .
| Target tissues | Actions |
|---|---|
| Cartilage | Inhibits terminal chondrocyte differentiation; increases chondrocyte proliferation |
| Bone | Maintains bone mass; promotes bone resorption during lactation |
| Mammary gland | Facilitates branching morphogenesis of mammary epithelium |
| Skin | Inhibits terminal differentiation of keratinocytes; promotes normal hair follicle development |
| Teeth | Promotes normal tooth eruption |
| Extraembryonic endoderm | Enhances the differentiation of primitive endoderm to parietal endoderm |
| Smooth muscle | Serves as a general smooth muscle relaxant |
| Central nervous system | Inhibits neuronal L-type calcium channel activity; protects neurons from excitotoxicity |
| Placenta | Maintains the positive maternal-fetal transplacental calcium gradient |
9.8.1
Endochondral bone development
The first direct evidence concerning a physiologic role for PTHrP appeared in 1994 with the report of the phenotype of mice lacking the expression of PTHrP due to targeted gene ablation . These animals died shortly after birth and were found to display a form of short-limbed dwarfism with generalized chondrodysplasia. The most striking feature of mice lacking expression of PTHrP is the disruption of normal endochondral ossification. Although the most obvious gross phenotypic abnormality is short-limbed dwarfism, the defect in endochondral bone formation is generalized.
The control of endochondral bone formation is maintained by a complex series of extracellular cues and intracellular signaling pathways . One of these factors is Indian hedgehog (Ihh), a member of the ancient hedgehog family of secreted patterning molecules. Ihh functions to promote chondrocyte proliferation and to maintain the pool of proliferating chondrocytes, thus extending the length of the differentiating cartilaginous growth plate prior to terminal differentiation and ossification . PTHrP appears to mediate some, but not all, of the actions of Ihh on endochondral bone formation . PTHrP directly inhibits the differentiation of proliferating chondrocytes to postmitotic prehypertrophic cells. Lack of PTHrP results in accelerated chondrocyte differentiation, shortened growth plates, and premature ossification. The cellular composition of the growth plates of PTHrP −/− animals is abnormal, with a marked reduction in the number of proliferating chondrocytes. Conversely, overexpression of PTHrP in chondrocytes of mice bearing a collagen II promoter-PTHrP transgene resulted in a distinct form of chondrodysplasia which is characterized by short-limbed dwarfism and delayed ossification . At birth, these animals displayed a cartilaginous endochondral skeleton, and histological evaluation revealed a marked suppression of the chondrocyte differentiation program. By 7 weeks of age ossification was evident, but the long bones remained foreshortened and misshapen. Similar abnormalities are seen in humans with hereditary Jansen’s metaphyseal chondrodysplasia, a disorder caused by mutations in the PTH/PTHrP receptor that result in constitutive receptor activation .
Ihh acts directly or indirectly on cells in the periarticular perichondrium to increase expression of the PTHrP gene . The effect of Ihh to delay terminal differentiation of chondrocytes in the long bones was not seen in PTHrP −/− or in PTH/PTHrP receptor −/− mice, indicating an intermediary role of PTHrP in Ihh action in endochondral bone formation. Consistent with this conclusion, a type II collagen promoter-driven constitutively active PTH/PTHrP receptor transgene rescues the abnormally accelerated chondrocyte differentiation program in Ihh −/− mice . These animals nonetheless displayed short-limbed dwarfism and decreased chondrocyte proliferation, demonstrating that PTHrP is not the only mediator of the multiple actions of Ihh on endochondral ossification. This conclusion is further supported by the observation that the severity of short-limbed dwarfism is much more severe in Ihh −/− , PTHrP −/− mice than in Ihh +/+ , and PTHrP −/− mice . It appears that chondrocyte differentiation is regulated in a complex fashion by these two secreted regulatory factors .
There is solid evidence that the PTH/PTHrP receptor plays an important role in initiating the actions of PTHrP on the differentiation of growth plate chondrocytes. The PTH/PTHrP receptor is expressed in proliferating chondrocytes as well as in cells in the transitional zone between proliferating and hypertrophic chondrocytes, where the regulation of terminal differentiation occurs . PTH/PTHrP receptor −/− mice display some of the growth plate abnormalities seen in PTHrP −/− mice . Interestingly, the deletion of the receptor does not fully recapitulate the deletion of PTHrP, raising the possibility that PTHrP may use signaling mechanisms in addition to the PTH/PTHrP receptor to regulate chondrogenesis. Patients with inherited mutations in the PTH/PTHrP receptor that cause constitutive (i.e., ligand-independent) signaling (Jansen metaphyseal chondrodysplasia) display growth plate abnormalities similar to those seen in mice overexpressing a collagen II promoter-PTHrP transgene . Lack of expression of functional PTH/PTHrP receptors in humans is associated with Blomstrand chondrodysplasia , a lethal disorder characterized by premature endochondral ossification. Recently, mutations in the PTHrP gene have been identified in patients with congenital limb deformities associated with autosomal-dominant brachydactyly type E .
9.8.2
Bone
There is considerable evidence that PTHrP plays a role in regulating skeletal homeostasis in skeletally mature mice. Mice with haploinsufficiency of PTHrP display no skeletal phenotype at birth but develop trabecular osteopenia after 3 months of age . These mice also show an increase in bone marrow adipose tissue, suggesting that reduced PTHrP expression favors adipogenic differentiation of mesenchymal progenitors. An osteopenic phenotype has also been observed in mice with targeted deletion of PTHrP expression in osteoblasts . This was associated with decreased recruitment of bone marrow stromal cell osteoblast precursors and increased apoptosis of osteoblasts. These findings suggest that production of PTHrP by cells of the osteoblast lineage plays a role in maintaining the pool of active osteoblasts that participate in bone formation. Expression of PTHrP appears to be required for normal formation of intramembranous as well as endochondral bone . The precise nature of the osteoblastic cells that express PTHrP is not clear, as PTHrP promoter activity was detected in a number of cell types in bone, but not in mature osteoblasts . PTHrP is also expressed in connective tissue cells in the outer layer of the periosteum and at sites of insertion of tendons and ligaments into cortical bone, and it is possible that PTHrP serves as a local regulator of bone formation or turnover in response to mechanical stimulation . More recently, a role for periosteal cell–derived PTHrP in fracture repair in mice has been demonstrated . Finally, deletion of PTHrP from osteocytes has been shown to reduce bone formation and osteoblast numbers . It thus appears that PTHrP produced at multiple skeletal sites contributes to skeletal anabolism.
9.8.3
Mammary gland
Targeted overexpression of PTHrP in mammary myoepithelial cells of transgenic mice provided direct evidence of a possible role for PTHrP in mammary gland development . The mammary ducts of 18- to 21-day-old transgenic mice were normal both in terms of the size of the ducts and in the branching morphogenesis of the developing gland. However, by 6 weeks of age, the transgenic animals displayed a delay in the development of the mammary duct system and a reduction in the degree of ductal branching. Pregnant transgenic animal displayed similar defects, as well as diminished formation of terminal ductules. Overexpression of PTH in mammary myoepithelial cells of transgenic mice produced identical morphogenetic defects, indicating that this action of PTHrP is mediated by the PTH/PTHrP receptor. The postnatal role of PTHrP in mammary gland development was studied in PTHrP −/− mice expressing a PTHrP transgene targeted to cartilage , allowing postnatal survival. At 4 months of age, female transgenic mice lack mammary glands. The mammary fat pads appear normal, but mammary epithelial ducts are missing. PTHrP −/− mice display the arrest of mammary duct development beginning between days 15 and 18 of embryogenesis. At this time, there is degeneration of epithelial elements within the ducts, and the initiation of normal branching morphogenesis of the mammary glands does not occur.
PTHrP is expressed in mammary epithelial cells , whereas functional PTH/PTHrP receptors are expressed in the underlying mesenchyme . This pattern of expression suggests that PTHrP is an epithelial signal that acts on PTH/PTHrP receptors in mesenchymal cells to promote mammary epithelial morphogenesis . Consistent with this notion, PTH/PTHrP receptor −/− mice display the same defects in embryonic mammary development seen in PTHrP −/− mice. Moreover, normal morphogenesis requires PTH/PTHrP receptor expression specifically in mammary mesenchymal cells . Humans lacking functional PTH/PTHrP receptors (Blomstrand chondrodysplasia) fail to develop nipples or breasts . The factors that regulate PTHrP expression during mammary morphogenesis are not well defined, but ectodysplasin A signaling through NF-κB has been implicated . Possible mesenchymal targets of PTH/PTHrP receptor signaling during breast development include the wnt pathway, BMP4, and transcription factors, including msx2 and lef1 . The mesenchymal genes encoding tenascin C and the androgen receptor are induced by PTHrP . PTHrP −/− or PTH/PTHrP receptor −/− male mice fail to display the normal androgen-dependent apoptotic destruction of the mammary bud, indicating that induction of the androgen receptor by PTHrP is essential for sexual dimorphism during mammary development. PTHrP production by mammary bud epithelial cells is also essential for the induction of nipple skin differentiation during mammary development .
A role for PTHrP during lactation was first suggested by the observation that suckling is a powerful stimulus for mammary PTHrP gene expression . Systemic maternal PTHrP levels were found to increase during suckling and to be elevated during lactation . These findings suggest that systemic PTHrP produced by the mammary gland may be important for mobilizing calcium destined for secretion into breast milk during periods of lactation. In support of this, mammary-specific deletion of the PTHrP gene in lactating mice was shown to reduce circulating levels of PTHrP and to attenuate bone loss during the lactation period . It appears that calcium mobilization during lactation results both from increased osteoclastic bone resorption and from increased osteocytic perilacunar remodeling (previously termed osteocytic osteolysis) . It is likely that both osteoclast-mediated bone resorption and osteocytic perilacunar remodeling contribute to PTHrP-mediated bone loss during lactation . Signaling by the CaSR in mammary epithelial cells downregulates mammary production of PTHrP , perhaps providing a mechanism for negative feedback in response to increased maternal levels of blood calcium. Interestingly, extremely large quantities of PTHrP are secreted into milk during lactation . Suckling animals and humans thus ingest large amounts of PTHrP over an extended time period, yet evidence that milk-derived PTHrP is absorbed in an active form and/or is physiologically important in suckling infants or animals is lacking.
9.8.4
Skin and teeth
Keratinocytes were the first normal cells shown to express PTH-like bioactivity and subsequently the PTHrP gene . PTHrP is expressed in the basal layer through the granulosa layer of the skin, with epidermal expression detectable as early as day 14 of embryogenesis in the rat , although PTHrP expression in the epidermis may be limited to the hair follicles . PTH/PTHrP receptors are present in dermal fibroblasts and keratinocytes , and novel binding sites for PTHrP have been detected in keratinocytes . In cultured human keratinocytes, suppression of PTHrP production resulted in increased cell proliferation and decreased differentiation . Thus PTHrP may have a role in the local regulation of epidermal cell proliferation and differentiation.
Targeted overexpression of PTHrP in basal keratinocytes and outer root sheath cells of hair follicles in transgenic mice resulted in a failure of ventral hair eruption that was evident within 6 days after birth . Dorsal hair was evident, but its eruption was delayed and the hairs were shorter and thinner compared to those of normal littermates. Histological evaluation of the transgenic mice revealed thickening of the ventral epidermis and expansion and increased cellularity of the dermis. Hair follicle development was substantially delayed in both ventral and dorsal skin of transgenic mice. These effects are probably due to the disruption of the normal epithelial–mesenchymal interactions required for proper hair follicle development and epidermal differentiation. PTHrP appears to promote anagen to catagen transition during the hair follicle cycle , and this may be mediated in part by an angiogenic action of PTHrP . PTHrP may also act earlier to facilitate entry into the anagen phase and initiation of the hair cycle .
PTHrP −/− mice that have been rescued by the expression of a type II collagen-PTHrP transgene display thinning of the epidermis with hypoplastic sebaceous glands and thinning of hair . These abnormalities could be reversed by the targeted expression of PTHrP in skin, indicating that PTHrP expression in basal keratinocytes is necessary for maintaining normal epithelial–mesenchymal interactions during epidermal differentiation. Inhibition of PTHrP action in skin was found to produce an increase in the number of follicles involved in active hair growth , and topical application of a PTH/PTHrP receptor antagonist stimulates hair growth in mice . These findings further support a role for PTHrP in regulating hair follicle development. PTHrP apparently maintains the pool of proliferating keratinocytes by suppressing their terminal differentiation, but the underlying mechanisms remain obscure.
PTHrP −/− mice display cranial chondrodystrophy with a failure in normal tooth eruption . PTHrP is expressed in the enamel epithelium, whereas the PTH/PTHrP receptor is expressed in the adjacent dental mesenchyme and in alveolar bone. These findings suggest that PTHrP is a regulator of epithelial–mesenchymal interactions during tooth development as well as promoting the resorption of alveolar bone that is required for normal tooth eruption. PTHrP increases the ratio of expression of RANKL:OPG by cementoblasts , an effect that presumably promotes the osteoclastic resorption required for tooth eruption . Exogenous administration of PTHrP to rats has been shown to promote tooth eruption . This effect is mediated by the PTH/PTHrP receptor, as humans lacking this receptor (Blomstrand chondrodysplasia) display a failure of tooth eruption . The deletion of the PTH/PTHrP receptor in mouse mesenchymal progenitor cells produces a similar phenotype .
9.8.5
Other actions of parathyroid hormone–related protein
PTHrP is expressed in a variety of smooth muscles where it functions as a local muscle relaxing agent. Increased intraluminal pressure (either from muscle contraction or from expanding intraluminal contents) is a known stimulus for PTHrP gene expression in the uterus and in vascular smooth muscle. Myometrial expression of PTHrP peaks just before the end of pregnancy, and this effect is specific for the pregnant uterine horn in unilaterally pregnant animals . Mechanotransduction is likely to be the primary stimulus, since physical stretch induces PTHrP expression in the nonpregnant rat uterus . Human amniotic fluid contains high levels of PTHrP , and it is possible that PTHrP produced in the amnion plays a role in suppressing myometrial contractions and/or in regulating chorionic blood flow. PTHrP is also expressed in the smooth muscle of the stomach, bladder, and oviduct, and promotes muscle relaxation in these tissues in response to distension . Pharmacological doses of PTH can reproduce the relaxing effects of PTHrP, strongly indicating the involvement of the PTH/PTHrP receptor.
PTHrP has effects on both the contractility and proliferation of vascular smooth muscle. PTHrP is widely expressed in vascular smooth muscle, and the administration of PTHrP in vivo and in vitro elicits vasodilatory responses in a variety of vascular beds . Expression of PTHrP in vascular smooth muscle is increased in experimental models of hypertension and in response to vasoconstrictors such as angiotensin II . Targeted overexpression of PTHrP in vascular smooth muscle of transgenic mice results in decreased baseline blood pressure as well as in a diminished hypotensive response to exogenous PTHrP, the latter possibly due to desensitization . The role of endogenous PTHrP is seen in transgenic mice overexpressing the PTH/PTHrP receptor in vascular smooth muscle . These animals are hypotensive, and (as expected) are hyperresponsive to exogenous PTHrP with respect to vasodilation. PTHrP appears to serve as an important physiologic regulator of static blood pressure and as a counter-regulatory factor secreted in response to vasoconstriction. PTHrP is expressed by endothelial cells , and this may contribute to the antiangiogenic effects of the protein. PTHrP is also induced in the blood vessels bathing skeletal muscle after muscle stimulation, perhaps promoting new capillary formation in response to increased muscle contraction .
As discussed above, PTHrP is expressed in the myometrium during pregnancy in response to distension produced by the growing fetus. By inducing relaxation of uterine smooth muscle, locally produced PTHrP permits progressive intrauterine growth of the fetus and may also assist in maintaining the uterus in a quiescent state until the onset of parturition. PTHrP also plays an important role in the fetal–placental unit during pregnancy . The protein is expressed in human amniotic tissue and may serve to increase chorionic blood flow . A role for fetal PTHrP in placental calcium transport is indicated by studies demonstrating that PTHrP −/− fetuses are hypocalcemic and have a reduced ability to accumulate calcium from the mother’s circulation . It appears that this action of PTHrP involves the midregion of the PTHrP molecule acting through a mechanism that does not involve the PTH/PTHrP receptor.
The genes encoding PTHrP and the PTH/PTHrP receptor are widely expressed in the central nervous system, with particularly high levels seen in cerebellar granule cells . These cells also express high levels of L-type calcium channels, and the expression of PTHrP appears to be induced by depolarization-induced calcium influx through these channels . Cerebellar granule cells are subject to excitatory cell death in response to agents such as kainic acid which trigger calcium entry through L-type calcium channels. PTHrP blocks this excitatory cell death by inhibiting L-type calcium channel activity through a mechanism that probably involves cAMP signaling via the PTH/PTHrP receptor . This is consistent with previous reports that exogenous PTH inhibits L-type calcium channel activity . These findings suggest that PTHrP functions as a neuronal survival factor produced in response to neuroexcitatory stimuli. Addition of a blocking antibody to PTHrP prevents cerebellar granule cell survival under depolarizing conditions, suggesting that PTHrP is the endogenous factor responsible for neuroprotection . Strong support for this concept is derived from studies in mice lacking the expression of PTHrP in the brain. Cortical neurons from these animals display a marked increase in sensitivity to kainic acid–induced excitotoxicity . PTHrP expression increases at sites of ischemic brain injury where it may play a protective role by enhancing blood flow . Recently, it has been suggested that PTHrP can promote heat and mechanical hypersensitivity by upregulating TRPV1 channels (capsaicin receptors) in dorsal root ganglion sensory neurons .
PTHrP may play a role in the maintenance of pancreatic β-cells. PTHrP and the PTH/PTHrP receptor are expressed in pancreatic β-cells, and overexpression of PTHrP stimulates β-cell proliferation and promotes improved glucose tolerance . Exogenous PTHrP is reported to increase β-cell regeneration in a mouse model of partial pancreatectomy .
9.9
Mechanism of action of parathyroid hormone and parathyroid hormone–related protein
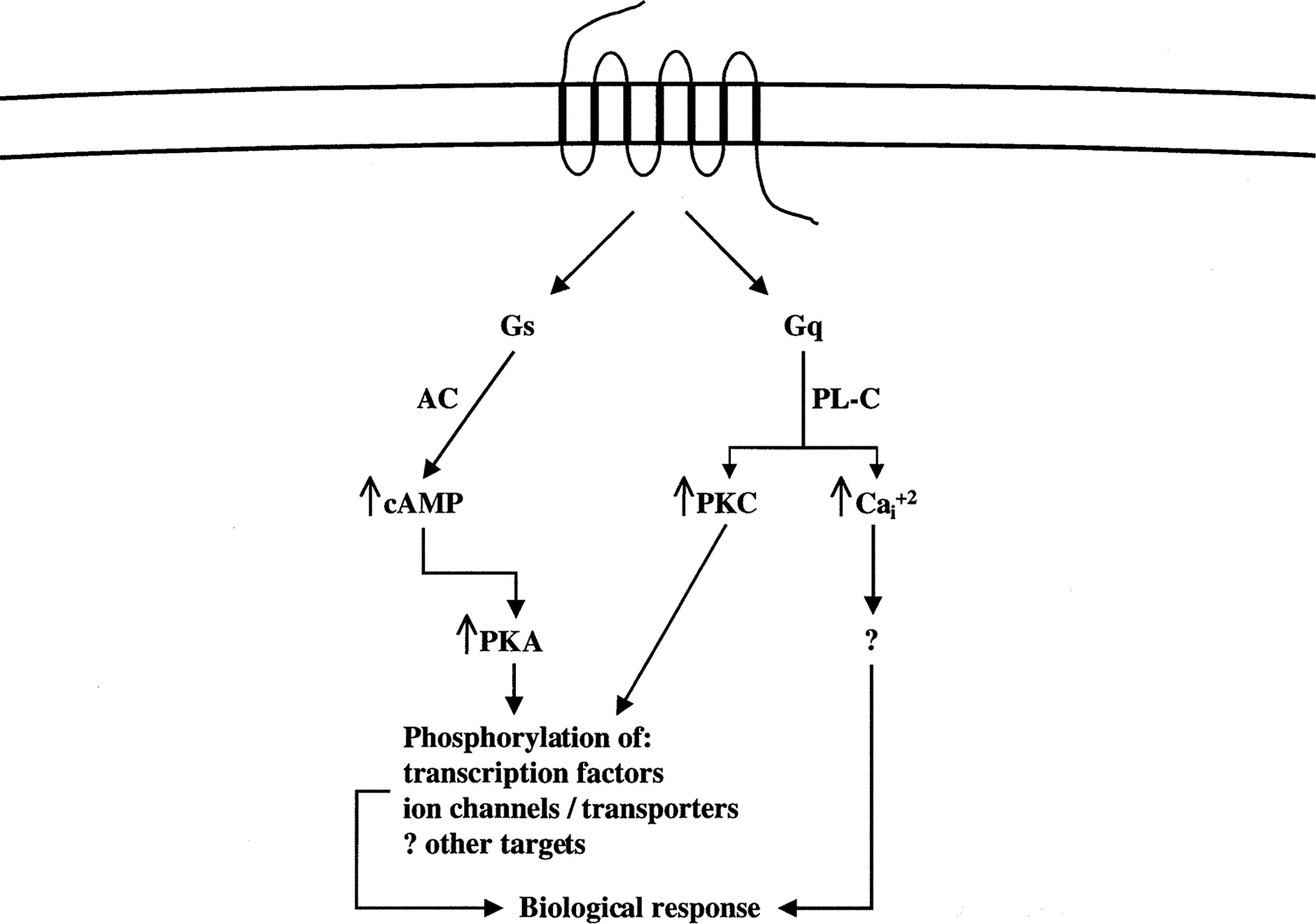
The actions of PTH and PTHrP are initiated by binding these proteins to the PTH/PTHrP receptor, a GPCR that predominantly activates two G proteins and thereby two major signal transduction pathways ( Fig. 9.5 ) . Shortly after the discovery of the cAMP signaling pathway, it was found that PTH is capable of increasing levels of cAMP in target cells through activation of the enzyme adenylyl cyclase . cAMP is a second messenger in the cellular action of a wide variety of hormones and other extracellular regulatory molecules. It activates cAMP-dependent protein kinase-A (PKA), which in turn phosphorylates and thereby regulates key proteins that participate in physiologic responses. The key substrates of PKA that are phosphorylated in response to PTH/PTHrP receptor activation include transcription factors, ion channels, transporters, and enzymes involved in cellular metabolism. PTH/PTHrP receptors also activate PLC, an enzyme that hydrolyzes the plasma membrane phospholipid phosphatidylinositol-4,5-bisphosphate to produce diacylglycerol (DG) and soluble 1,4,5-inositol trisphosphate (IP 3 ). DG and IP 3 function as second messengers—the former by activating protein kinase-C (PKC), and the latter by binding to and opening calcium channels on the membrane of the endoplasmic reticulum—thereby increasing cytosolic free calcium.
Related posts:
 Osteoclast biology
Osteoclast biology
 Impact of physical characteristics and lifestyle factors on bone density and fractures
Impact of physical characteristics and lifestyle factors on bone density and fractures
 Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
 Relationship between periodontal disease, tooth loss, and osteoporosis
Relationship between periodontal disease, tooth loss, and osteoporosis
 On the evolution and contemporary roles of bone remodeling
On the evolution and contemporary roles of bone remodeling
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


