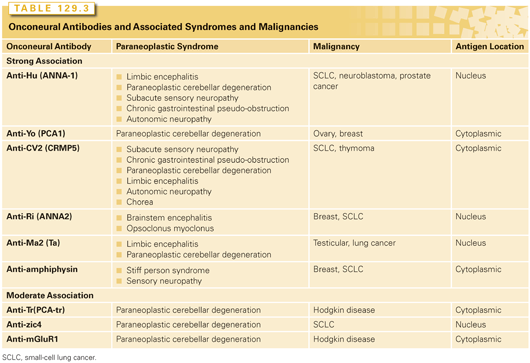
Frequently, a panel of serum antibodies against neural proteins, referred to as onconeural antibodies, are checked as part of the initial workup, as a single syndrome may be associated with multiple antibodies.10 These antibodies are useful but have some well-recognized flaws. The association between onconeural antibodies and PNS is variable (Table 129.3).9 Onconeural antibodies found in both PNS and nonmalignant syndromes include antiacetyl-choline receptor, antinicotinic, antivoltage-gated calcium channel (VGCC), and voltage-gated potassium channels (VGKC).1,4 Among the known VGCC subtypes including L, R, T, N, and P/Q,11,12 the latter two are expressed in SCLC and are common targets for antibodies in these patients.13 All these antibodies target extracellular antigens. Importantly, <50% of patients with a clinical syndrome of PNS will have the antibodies detected, and these antibodies can be detected in patients without PNS.1,10 Therefore, the absence of onconeural antibodies should not exclude the diagnosis of PNS, and the presence of onconeural antibodies alone should not be considered diagnostic. Of note, anti-Tr antibodies associated with cerebellar degeneration can be detected in the CSF but not in the serum.14 Another clinical dilemma is that onco-neural antibody tests take time and symptomatic patients may require treatment before the results are available.
Patients may present with symptoms consistent with PNS without a known diagnosis of cancer, and a workup for an underlying malignancy is initiated. This should include a history to assess for symptoms and risk factors for an underlying malignancy, a physical exam, and a workup for occult malignancy. Frequently, the initial test is computed tomography scan of the chest, abdomen, and pelvis. Fluorodeoxyglucose positron emission tomography (FDG-PET) is increasingly popular. A small study compared computed tomography and FDG-PET, and the sensitivity for detection of tumor or tumor recurrence in patients with PNS was 30% and 90%, respectively (p <0.01).15
Specific Syndromes
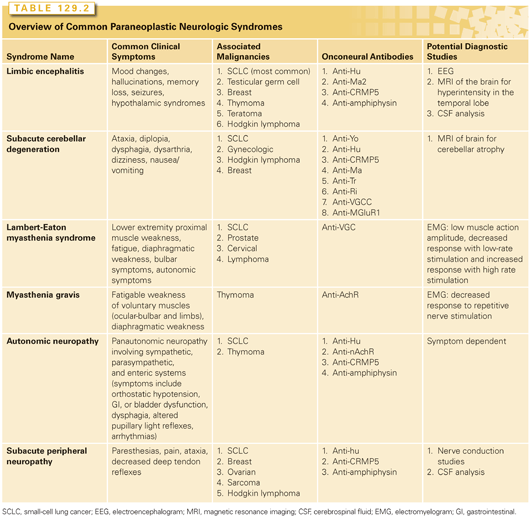
Numerous neurologic syndromes have reported to be associated with malignancy in case reports. However, several specific syndromes with a common constellation of symptoms have been reported consistently in the literature. This section will focus on the more common PNSs. PNSs that affect the central nervous system include LE, subacute cerebellar degeneration, and OMS. Symptoms associated with LE include short-term memory loss, confusion, psychiatric symptoms such as hallucinations and mood changes, and seizures. The symptoms will present over a period of several days to several months. Magnetic resonance imaging (MRI) reveals hyperintensity of the medial temporal lobe(s), and CSF analysis will reveal pleocytosis, elevated protein, elevated IgG, and oligoclonal bands.1 LE is associated with onconeural antibodies against the VGKC.9 Subacute cerebellar degeneration typically presents with symptoms of ataxia, diplopia, dysphagia, and dysarthria. Typically, the first symptoms are isolated gait ataxia and subsequent development of truncal and hemispheric cerebellar dysfunction.9 The onset of symptoms is generally <12 weeks, and MRI of the brain does not reveal any significant cerebellar atrophy. The P/Q type anti-VGCC antibodies and other antibodies have been reported with this PNS.1,16 Concurrent cerebellar degeneration and LEMS has been reported as well in patients with SCLC.17 OMS is characterized subacute onset of opsoclonus, a disorder or saccadic eye movements, chaotic saccades in all directions, and arrhythmic-action myoclonus that predominately involves the trunk, limbs, and head.18 The pathophysiologic mechanism for this disorder is unclear but may be mediated by disruption of the Nova-1/Nova-2 proteins of the glycine receptor.19 This disorder has been associated with SCLC in adults and neuroblastoma in children.19
Subacute sensory neuropathy and GI dysmotility are two PNSs that affect the peripheral nervous system. The common symptoms of subacute sensory neuropathy are numbness and pain of the extremities, marked asymmetry of symptoms at onset, and loss of proprioception; the onset symptoms is often <12 weeks.1,9 In the later stages, patients can develop ataxia and multifocal involvement. On physical exam, there is loss of the deep tendon reflexes, and electrophysiologic studies reveal involvement of sensory fibers and absence of sensory nerve action potentials.
Patients with cancer often report GI symptoms, including anorexia, early satiety, nausea, emesis, abdominal pain, diarrhea, or constipation. Among the multiple causes for GI complaints in patients with cancer is a paraneoplastic dysmotility, which may affect either the entire GI tract or isolated segments, where they produce specific syndromes such as achalasia, gastroparesis, and intestinal dysmotility with constipation.20 Paraneoplastic dysmotility should be suspected in patients with rapid symptom onset, significant weight loss, presence of concurrent autonomic or sensory neuropathy, and risk factors for malignancy such as smoking and strong family history. The onset of GI dysmotility usually precedes the diagnosis of cancer, and the most common associated malignancy is SCLC. The presumed pathophysiology is the presence of tumor antigens that elicit an immune response which cross-reacts with the enteric nervous system. The most common antibody associated with GI dysmotility is the antineuronal nuclear antibody-1, also known as anti-Hu, and approximately 30% of patient with this antibody have symptoms of GI dysmotility.20,21 Other antibodies associated with GI dysmotility include antineuronal nuclear antibody-2 (anti-Ri), VGCC types N and P/Q, VGKC, and Purkinje cell cytoplasmic (anti-Yo). In a retrospective evaluation of 12 patients diagnosed with malignant tumors and GI motor dysfunction, SCLC was the most common primary malignancy, accounting for 9 cases.22 The three other patients had lung adenocarcinoma, follicular lymphoma, and ovarian papillary serous adenocarcinoma. The most common presenting symptoms were weight loss, nausea, emesis, and early satiety, which were acute in onset and rapidly progressive in all patients. Although the symptoms preceded the diagnosis of SCLC by a mean interval of approximately 9 months, they developed after the diagnosis in patients with other tumors. Among the 11 patients with serum available, 10 had one or more markers of paraneoplastic immunity including antineuronal nuclear antibody-1, which was detected in all 8 patients with SCLC tested.
MG and LEMS are two PNSs that impact the function of the neuromuscular junction. MG is characterized by muscle weakness of the voluntary muscles that is worsened by exertion. Common symptoms are diplopia and ptosis related to eye muscle weakness. Other symptoms include dysarthria, problems with chewing and swallowing caused by oropharyngeal weakness, and limb girdle weakness, which is generally more pronounced in the proximal than distal muscle groups. Approximately 85% of patients have an onconeural antibody against the muscle acetylcholine receptor (antiacetylcholine receptor), and about 6% of patients will have an onconeural antibody against the muscle specific kinase known as “striational antibodies.”23 MG is associated with thymoma. MG occurs in 30% to 50% of patients with thymoma but only 15% of patients with MG will have thymoma. The presence of antistriational antibodies in a patient younger than 60 years is suspicious for a thymoma.23 Patients with MG and thymoma may have a better prognosis due to early detection and/or more indolent disease.24 Importantly, thymectomy will result in an improvement of the symptoms of in the MG, and all thymic tissue should be removed to reduce the rate of recurrence.25
Approximately 50% to 60% of patients with LEMS have an underlying tumor, the most commonly SCLC.26 Clinical symptoms include proximal muscle weakness, autonomic features such as dry mouth, constipation, erectile dysfunction, and less commonly orthostatic hypotension and micturition difficulties, and reduced tendon reflexes. Proximal muscle weakness is frequently the first symptom.26 Distinguishing MG from LEMS on clinical symptoms can be difficult. In MG, the initial weakness frequently affects the extraocular or bulbar muscles, whereas in LEMS the proximal muscle weakness is frequently the presenting symptom.27 Patients with LEMS frequently present with leg weakness, whereas limb weakness restricted to the arms is more common in patients with MG. Onconeural antibodies against the P/Q-type VGCC on the presynaptic nerve terminal are characteristic of LEMS and the cause of the muscle weakness. Electromyography studies reveal a low amplitude compound muscle action potential, which becomes lower at low-stimulating frequencies.26 In order to distinguish LEMS from MG, high-frequency or postexercise stimulation are the diagnostic tests. An increased compound muscle action potential of 60% is considered abnormal and is very sensitive and specific for LEMS.28 Postexercise stimulation and high-frequency stimulation have a similar sensitivity, but high-frequency stimulation is painful and should be avoided if possible.26
Treatment
Given the rarity of PNS, there are no “evidence-based” studies for optimal care. In general, care follows the principles of treating the underlying malignancy, immunosuppression, and symptom-directed supportive care. Many times, the care of a patient with a PNS requires the coordination of care with oncologists, neurologists, and specialists in palliative care. The appropriate care for the type and stage of underlying malignancy should be initiated. Immunosuppression used in the treatment of PNS including corticosteroids, alkylating agents such as cyclophosphamide, intravenous Ig (IVIG), rituximab, and plasmaphaeresis. The prednisone dose frequently employed is 1 mg/kg daily or high-dose intravenous methylprednisolone (1 gm/day for 5 days, followed by weekly therapy of the same dose for 6 to 12 weeks).10 The dose of oral cyclophosphamide frequently used is 2 mg/kg daily, and azathioprine dose frequently used is up to 2.5 mg/kg daily with adjustments related to toxicity such as myelosuppression.
IVIG is believed to modulate the immune system by interacting with the Fc receptor on immune cells, neutralizing the autoimmune antibody and/or increasing the clearance of the autoimmune antibody, and increasing the regulatory T-cells.29 PNSs that frequently respond to IVIG include LEMS, MG, Guillain-Barre syndrome, stiff person syndrome, and anti-VGKC limbic encephalitis.29 IVIG is frequently given 400 to 1,000 mg/day for a total of 2 to 3 g, and the most serious toxicity is nephrotoxicity.29 Plasmapheresis removes the onconeural antibodies from circulation, and an improvement in symptoms can be observed within days. The effect generally only lasts several weeks, and the concurrent administration of immunosuppressive drugs is required.1 Rituximab, an anti–B-cell monoclonal antibody, has been used as immunosuppression and case reports of efficacy exist in the treatment of OMS, stiff-person syndrome, paraneoplastic cerebellar degeneration, and LE.30,31 Rituximab has been mainly studied in patients with PNS who demonstrate the presence of an established onconeural antibody, and the rituximab did not result in consistent effect of serum or CSF antibody titers.31 The standard dose of rituximab of 375 mg/m2 weekly for 4 weeks has been used.
Some PNS benefit from therapies focused on reducing symptoms. The best examples of this are LEMS and MG in which treatment was with the cholinesterase inhibitor pyridostigmine, which inhibitors the degradation of acetylcholine at the neuromuscular juncture and may improve muscle strength.32 For patients with LEMS, guanidine inhibits the VGKC and enhances the release of acetylcholine. However, the toxicities of myelosuppression and renal insufficiency make the use of this agent problematic, especially if the patient requires cytotoxic chemotherapy.33 Aminopyridines prolong nerve terminal depolarization, which increases calcium and consequently the release of acetylcholine. Whereas dalfampridine, the first available aminopyridine, is not routinely used due to toxicity concerns, 3,4-diaminopyridine is better tolerated and has been shown to improve motor and autonomic symptoms.34,35 The common adverse effects include lightheadness, fatigue, epigastric distress, and difficulty sleeping. For patients with autonomic neuropathy and symptomatic hypotension, treatment options include increased water intake, fludrocortisone, midodrine, and caffeine.36,37 For patients with pseudo-obstruction, neostigmine has been investigated.38
Prognosis
Given the rarity of and the multiple different malignancies associated with this PNS, it has been difficult to determine the impact of this PNS of prognosis. In the case of MG associated with thymoma, there is some evidence that patients may have a better prognosis due to more indolent disease and/or earlier detection, and for patients with SCLC with anti-Hu antibody–associated PNS, there is some evidence that they are more responsive to therapy.24,39 Conversely, patients with a PNS often have poor performance as a consequence of well-advanced malignancy, and many times there are justifiable concerns about the patient’s ability to tolerate cytotoxic chemotherapy. If the poor performance status is related to the underlying PNS, cancer-specific therapy is often initiated in an attempt to improve the symptoms. Although the exact impact of a concurrent PNS on prognosis is difficult to assess, survival consistent with the prognosis of the underlying malignancy has been reported with treatment.
PARANEOPLASTIC ENDOCRINOLOGY SYNDROMES
Inappropriate Antidiuretic Hormone Secretion
The syndrome of inappropriate antidiuretic hormone secretion (SIADH) is a disorder of sodium and water balance characterized by a hypotonic euvolemia hyponatremia. The nonapeptide antidiuretic hormone (ADH) or arginine-vasopressin is synthetized in the hypothalamic magnocellullar neurons of the supraoptic and paraventricular nuclei and stored in the posterior lobe of the pituitary gland. There are three known receptors for ADH at the cell membrane: V1a, V1b, and V2.40,41 V1a receptors are found in the vascular smooth muscle, uterus, platelets, and hepatocytes, where the main functions include vasoconstriction, uterine contraction, platelet aggregation, and glycogenolysis, respectively. V1b receptors are located in the corticotroph cells of the anterior pituitary gland where they are involved in the release of adrenocorticotrophin hormone (ACTH) and beta-endorphin. V2 receptors are located in the basolateral membrane of the renal-collecting duct principal cells, vascular smooth muscle, and vascular endothelium, where they are involved in the synthesis and insertion of aquaporin 2 (AQP-2) channels into the apical membrane, vasodilation, and release of von Willebrand and factor VIII. Binding of ADH to the V2 receptor in the basolateral membrane of the collecting duct cells leads to activation of adenylate cyclase, with increased production of cyclic adenosine monophosphate, which in turn leads to activation of protein kinase A. Protein kinase A phosphorylates vesicles containing preformed AQP-2 vesicles, which leads to their movement to the apical membrane, where AQP-2 is inserted and works as selective water-conducting channel according to the gradient from the tubular lumen to the medullary interstitium. V2 receptor activation also leads to the increased synthesis of AQP-2. The water enters the cells through AQP-2 channels in the apical membrane and exits through the constitutively active AQP-3 and AQP-4 in the basolateral membrane. The two main stimuli for the ADH secretion are the increased plasma osmolality and decreased intravascular volume, which are regulated by the hypothalamic central sensing system and baroreceptors, respectively. Both ADH and thirst maintain the serum osmolality within a narrow range averaging approximately 285 mOsm/kg, with an increased osmolality above the threshold being the most important stimulus for the ADH secretion. A second stimulus is a decrease in the effective arterial volume caused by extracellular volume depletion or hypotension, which is detected by the volume-sensitive receptors on the left atrium and leads to a restoration of the effective blood volume through V2 receptors and the blood pressure through V1A receptors in the vascular smooth muscle.42 Stimulation of V1a receptors occur at higher plasma concentrations of ADH compared those needed for V2-dependent antidiuretic effects.
The main etiologies for SIADH can be broadly categorized into malignancies or drug-induced and pulmonary or central nervous system disorders. Both intrathoracic and extrathoracic malignancies have been associated with SIADH, with SCLC usually identified as the most common cause. List and colleagues43 reported the presence of SIADH in 40 (11%) out of 350 patients with SCLC, without correlation to stage or site of metastases, and no influence, as an independent variable, on response to chemotherapy or survival. SIADH has been described in approximately 3% of patients with head and neck cancer,44 most commonly in tumors of the oral cavity.45 Other malignancies that have been rarely associated with SIADH include thymomas,46,47 lymphoma,48–50 pancreatic carcinomas,51 and prostate cancer.52–54
Clinical Manifestations
The symptoms from SIADH depend on the degree and rapidity of the change in the serum sodium levels, with the rate of sodium fall considered to be a stronger predictor for morbidity and mortality compared its magnitude.55,56 Severe hyponatremia is arbitrarily defined as serum sodium concentration <115 mmol/L.57 Due to hypoosmolality, there is an entry of free water into the cells, with the brain being particularly affected as it has limited space for expansion inside the skull. Therefore, the most common symptoms from hyponatremia are neurologic and result from cerebral edema. The presence of brain edema triggers an adaptive response to restore the brain volume and limit the neurologic complications, with an initial decrease in brain electrolyte content followed by loss of organic solutes such as creatine and amino acids.58 During acute hyponatremia, defined as the rapid decline of serum sodium over a period of <48 hours, the adaptive brain response is overwhelmed, with the development of severe brain edema and neurologic symptoms including nausea and vomiting and headaches, which may progress to seizures, respiratory arrest, and death. Patients with chronic hyponatremia are usually either asymptomatic or have milder symptoms compared to acute hyponatremia, including fatigue, confusion, falls, and attention deficit.59
Diagnosis
The diagnostic criteria for SIADH may be subdivided into essential and supplementary features (Table 129.4).60–62 The diagnosis of SIADH is usually made after exclusion of other etiologies for hyponatremia through the use of the essential diagnostic criteria. The first step is to measure the plasma osmolality, with values <275 mOsm/kg of water ruling out the causes of hyponatremia with normal or increased plasma osmolality such as translocation by hyperglycemia or pseudohyponatremia caused by hyperlipidemia or hyperproteinemia. The next step is to evaluate the urinary osmolality, which should be >100 mOsm/kg of water during hypotonicity, to define the presence of inappropriate renal dilution and help to exclude the few causes of hyponatremia with decreased urinary osmolality such as beer potomania and polydipsia. Clinical euvolemia is defined as the absence of signs of volume depletion (dry mucous membranes, tachycardia, and orthostasis) caused by renal or extrarenal sodium loss or volume overload (edema and ascites) caused by renal failure, nephrotic syndrome, cirrhosis, and heart failure. The next step is to rule out hypothyroidism and glucocorticoid deficiency, the other two common etiologies that fall into the hypotonic euvolemic hyponatremia.

The supplementary features include reduced uric acid and blood urea nitrogen, increased fractional excretion of sodium and urea, correction of hyponatremia with fluid restriction but not after a 2 liter infusion of 0.9% saline, elevated plasma ADH levels despite hypotonicity and clinical euvolemia and abnormal result on the test of water load, which is defined as inability to excrete at least 90% of a 20 mg/Kg water load in 4 hours or failure to achieve an urine osmolality <100 mOsmol per Kg of water. Decreased blood urea nitrogen and uric acid are both indicative of euvolemia although not specific for SIADH, whereas increased values are suggestive of hypovolemia. Because some patients with nonparaneoplastic SIADH may have low or undetectable levels of ADH, the term syndrome of inappropriate diuresis has been proposed as a more accurate description of this condition.63
Treatment
The treatment of paraneoplastic SIADH may be broadly subdivided into indirect and direct modalities. In addition to cancer-specific therapy that, if effective, may eventually improve the hyponatremia by reducing the production of ADH by the malignant cells, the initial treatment for nonsevere cases of hyponatremia caused by SIADH is fluid restriction to approximately 800 ml daily in an attempt to induce a negative balance as the urinary excretion plus insensible losses usually exceed 1 L per day.55 This treatment modality, however, is associated with a slow improvement, has a very low compliance rate, particularly in outpatients, and is often insufficient to improve the hyponatremic symptoms. Therefore, pharmacologic interventions are usually indicated. Oral administration of urea at 30 g daily has been associated with improvement in hyponatremia associated with SIADH by causing osmotic diuresis and increased water excretion.64–66 The main drawback from urea is the poor palatability, with most patients unable to tolerate it despite being dissolved in strong flavored liquids. Loop diuretics such as furosemide and triamterene have been shown to be effective in patients with SIADH by causing increased excretion of free water.67 Both demeclocycline and lithium decrease the responsiveness to ADH in the collecting tube, causing nephrogenic diabetes insipidus, with decreased urine concentration despite elevated ADH.68–71 Demeclocycline is a tetracycline derivative with unpredictable onset of action and may cause significant side effects including GI intolerance, photosensitivity, and azotemia. The typical doses are from 300 to 600 mg twice daily. Lithium improves hyponatremia through dysregulation of AQP-272 but has been abandoned as a treatment for SIADH due to the association with interstitial nephritis and end-stage renal failure as well as decreased efficacy compared to demeclocycline.69
Patients with symptomatic acute hyponatremia should be treated with continuous infusion of hypertonic saline (3% NaCl), as the euvolemic hyponatremia is unlikely to respond to normal saline, with a maximal correction rate of 8 to 12 mmol/L during the first 24 hours.62 The initial infusion rate can be estimated by multiplying the patient’s body weight in kilograms by the desired rate of increase of serum sodium in mmol/L per hour.73 As an example, for a 70-kg patient, an infusion of 3% saline at 70 ml and 35 ml/hour will increase the serum sodium by approximately 1 mmol/L and 0.5 mmol/L per hour, respectively. Patients with known cardiovascular disease should receive 20 to 40 mg of intravenous furosemide to avoid volume overload. The main goal of therapy is to achieve a safe serum levels with symptomatic relief, rather than completely normal levels. Therefore, acute treatment can be discontinued once the symptoms are resolved, a safe serum sodium usually defined as ≥120 mmol/L, or the total magnitude of correction reached 18 mmol/L.
Vasopressine Receptor Antagonists
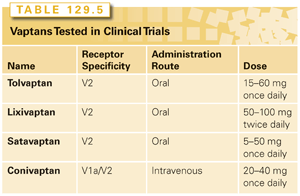
Vasopressine receptor antagonists (vaptans) are nonpeptide ADH (arginine-vasopressin) antagonists that penetrate deeper into the transmembrane region of V2 receptors compared to ADH, and competitively block the activation of the receptor by endogenous ADH.74 With the inactivation of the receptor, the synthesis and transport of AQP-2 into the apical membrane of the collecting duct cells is inhibited, resulting in a decrease in the free water reabsorption and increased urine volume.75 Although the increased diuresis produced by V2 receptor antagonists is quantitatively similar to loop diuretics, the latter produces essentially an aquaresis, which unlike the diuretics, is characterized by decreased urinary osmolality. Vaptans are indicated for patients with either euvolemic or hypervolemic hyponatremia, and should not be used in hypovolemic hyponatremia. During treatment with vaptans, it is very important to monitor the sodium levels to exclude rapid correction rates; there is no need for fluid restriction. Among the four vaptans with reported data from randomized clinical trials, only conivaptan and tolvaptan are currently approved by the US Food and Drug Administration and are available for clinical use (Table 129.5). Conivaptan is the only drug that inhibits both V2 and V1a receptors, while the other vaptans are V2 receptor–specific.76–78 Another difference between conivaptan and the other vaptans is that although the former was originally developed as an oral preparation,79,80 it was subsequently approved by the US Food and Drug Administration and marketed as an intravenous drug.81 The recommended treatment includes a loading dose of 20 mg over 30 minutes followed by a maintenance dose of 20 to 40 mg daily through continuous infusion for up to 4 days. Due to infusion site complications, including erythema, phlebitis, and swelling, the infusion should be through a large vein with the site changed every 24 hours. There are several medications with potential for significant drug interactions including statins, calcium channel blockers, benzodiazepines, azole antifungals, and chemotherapy agents including vinca alkaloids, doxorubicin, and etoposide, with the latter particularly important given the frequency of SCLC as the SIADH etiology, for which etoposide is commonly the chemotherapy of choice in the first-line setting. Tolvaptan is an oral drug that is 29 times more selective for V2 receptors than V1a and does not have intrinsic agonist effects.82,83 Schrier and colleagues84 reported the combined results of two multicenter randomized clinical trials, the Study of Ascending Levels of Tolvaptan in Hyponatremia (SALT) 1 and 2, where 448 patients were randomized to oral tovaptan 15 mg daily or oral placebo. During the first 4 days of therapy, the dose of the study drug could be escalated to up to 60 mg daily according to the serum sodium concentrations. Eligible patients had hyponatremia due to cirrhosis, congestive heart failure, or SIADH. Tolvaptan was associated with a significant increase in the serum levels of sodium during the first 4 days (p <0.001) and after 30 days of therapy (p <0.001). The treatment was well tolerated with increased thirst, dry mouth, and urination as the main side effects. In a subset analysis of 110 patients with SIADH from the SALT trials, the results were similar to the overall population, with a significant improvement in the serum sodium concentration both in the first 4 days and on day 30.85 The long-term administration of tolvaptan was evaluated by the open-label SALTWATER study, which was an extension of the SALT studies where 111 patients previously assigned to both placebo and tolvaptan arm received tovalptan for an extended period of time.86 Prolonged treatment was associated with maintenance of normal serum sodium levels and tolerable profile where the most common toxicities were similar to SALT studies, including dry mouth, thirst, polydipsia, and polyuria. The most likely role for vaptans in SIADH is in the treatment of mild to moderate hyponatremia and severe asymptomatic hyponatremia.87 There is not enough data to support the use of vaptans in patients with symptomatic hyponatremia, who should be treated with hypertonic saline.
One of the main concerns during the treatment of chronic hyponatremia is the overzealous treatment, which may lead to osmotic demyelination.88–90 This complication, also known as osmotic demyelination syndrome (ODS), occurs when the hyponatremia is corrected too rapidly, outpacing the recapture of lost organic molecules by the brain. During the rapid increase in the serum osmolality, the brain cells try to reverse the loss of solutes by increasing the production of organic osmolytes and inorganic ions.91 This process, however, causes a significant metabolic strain on glial cells leading to adenosine triphosphate depletion. The shifting of water to outside the cells during the correction of hyponatremia results in shrinkage of the glial cells, cellular damage, and disruption of the blood–brain barrier, with the latter allowing inflammatory mediators to enter the central nervous system where they may cause damage to the oligodendrocytes and demyelination. Some central nervous system areas such as the pons appear to be more vulnerable than others. The main risk factors for the development of ODS are serum sodium ≤105 mmol/L, hypokalemia, alcoholism, malnutrition, and advanced liver disease. ODS usually follows a biphasic course with an initial symptomatic improvement with the correction of the hyponatremia, which is followed by new and progressive neurologic symptoms developing a few days later. The presentation symptoms range from confusion to progressive quadriplegia associated with dysphagia and dysarthria. ODS is best evaluated by MRI where hypodense nonenhancing lesions can be seen on T1-weighted images and hyperdense lesions on T2-weighted images.92,93
Ectopic Cushing Syndrome
Cushing syndrome is a constellation of symptoms caused by a sustained exposure to excessive concentrations of circulating glucocorticoids. The most common cause of Cushing syndrome is iatrogenic, due to exogenous administration of glucocorticoids. Endogenous Cushing syndrome may be subdivided into ACTH (or corticotropin) dependent and independent.94 ACTH-dependent Cushing syndrome is far more common, accounting for approximately 80% to 85% of the cases and subdivided into Cushing disease, which is caused by pituitary adenomas and accounts for 80% of the ACTH-dependent endocrine causes, ectopic secretion of ACTH by nonpituitary tumors in approximately 20% of cases, and the rare ectopic secretion of corticotropin-releasing hormone (CRH). ACTH-independent Cushing syndrome is almost always caused by adrenal adenomas or carcinomas, with <1% of cases caused by macronodular adrenal hyperplasia, primary pigmented nodular adrenal disease, and McCune-Albright syndrome.
The most common tumors associated with ectopic ACTH production are the SCLC and bronchial carcinoids, with each accounting for approximately 22% of cases in the largest series.95–99 Other reported tumors include GI neuroendocrine tumors, thymic carcinoids, medullary thyroid carcinoma, and pheochromocytoma. Occult tumors have been reported in approximately 13% of cases, ranging from 6.7% to 18.9%. In a retrospective review of 545 patients with SCLC seen at the Toronto General Hospital between 1980 and 1990, there were 23 patients (4.5%) with Cushing syndrome caused by ectopic ACTH production. These patients had low response to chemotherapy, high rate of complications from the chemotherapy, and shorter survival.
The clinical features of Cushing syndrome are variable, with decreased libido, obesity or weight gain, facial plethora or rounding, menstrual changes, hypertension, and hirsutism as the most common manifestations.100 The characteristic features of increased supraclavicular fat, proximal muscle weakness, and purple striae occur in a minority of patients. When compared to neuroendocrine tumors, SCLC has been shown to be associated with higher frequency of hyperpigmentation and ankle edema, but lower frequency of psychiatric findings.98 Due to the high percentage of SCLC and other advanced malignancies, which are usually associated with weight loss, weight gain is less common in paraneoplastic than nonmalignant Cushing syndrome.
Diagnosis
Patient with characteristics of glucocorticoid excess should have a biochemical confirmed diagnosis of hypercortisolemic state prior to proceeding to the differential diagnosis of Cushing syndrome.
The initial screening tests for Cushing syndrome are the 24-hour urinary free cortisol, low-dose dexamethasone suppression, and the late-night salivary cortisol tests.101–103 The measurement of urinary cortisol allows a direct assessment of the circulating free cortisol. In contrast, plasma cortisol levels measure the total cortisol levels, including both free and bound to cortisol-binding globulin or other serum proteins. When the excess of cortisol saturates the serum-binding proteins, it is excreted in the urine in a free or unbound form. Therefore, unlike serum cortisol, urinary levels measure only the biologically active form and are not affected by conditions and drugs that alter cortisol-binding globulin. Urinary free cortisol values higher than four-fold greater than the upper normal limit are rare in disorders other than Cushing syndrome. However, up to three 24 hour collections may be required to exclude intermittent hypercortisolism, and the test is not reliable in patients with creatinine clearance <30 ml/min.
There are two low-dose dexamethasone suppression tests commonly used, based on the fact that in normal subjects, the administration of supraphysiologic doses of glucocorticoids suppresses ACTH and cortisol secretion.104 In patients with endogenous Cushing syndrome, however, the administration of low-dose dexamethasone does not suppress ACTH. The overnight test involves the administration of 1 mg of dexamethasone between 11 p.m. and 12 a.m. with measurement of fasting serum cortisol between 8 and 9 a.m. of the following day. In the 48-hour test, patients receive 0.5 mg of dexamethasone every 6 hours for eight doses, starting at 9 am on day 1, with serum cortisol measured at the start of the test and 6 hours after the last dose. Serum cortisol levels <1.8 mcg/dl have a sensitivity and specificity rates of >95% and 80%, respectively, in excluding Cushing disease. The late-night salivary cortisol test is based on the high correlation between free serum and salivary cortisol levels, independently of salivary flow rates.105 Furthermore, unlike the serum counterpart, salivary cortisol is not significantly affected by changes in the serum-binding proteins. Due to its easy collection and prolonged stability at room temperature, the late-night salivary cortisol test appears particularly suitable for outpatient screening of Cushing disease. The saliva samples are collected during expected plasma cortisol nadir, between 11 p.m. and 12 a.m., via a cotton swab or by spitting into a tube, which can be sealed and transported at room temperature. Normal reference levels are dependent on the assays used, with cutoff values used in trials ranging from 3.6 to 15.2 mmol/L. Therefore, the values should be validated for each laboratory. The presence of normal salivary cortisol levels from samples obtained in two separate days makes Cushing syndrome very unlikely. The diagnosis of Cushing syndrome is confirmed when two tests are unequivocally abnormal, with additional evaluation required in case of slightly abnormal or discordant test results.
Once the diagnosis of Cushing disease is confirmed, the next step is to establish the cause among the three broad subgroups including the ACTH-independent adrenal etiologies, ACTH-dependent pituitary, and ectopic causes. The workup starts with the measurement of plasma ACTH, with levels <5 pg/mL and >20 pg/mL indicating ACTH independent and dependent, respectively.106–108 In cases with intermediate values, the CRH stimulation test may help to clarify the ACTH dependency.109 In this test, 100 mcg of CRH is administered intravenously with measurements of ACTH before and after the infusion. Increase in ACTH suggests ACTH-dependent causes. ACTH responses, however, are more common in Cushing disease than ectopic ACTH secretion. As both pituitary and ectopic causes belong to the ACTH-dependent category, the next task is to rule out Cushing disease, usually by a brain MRI but occasionally with bilateral inferior petrosal sinus sampling, which is considered the gold standard for the differential diagnosis of ACTH-dependent Cushing syndrome.110–113 In this test, ACTH is collected simultaneously from both inferior petrosal sinuses, which drain directly from the pituitary circulation and blood. CRH stimulation is occasionally used to increase the test sensitivity. Petrosal sinus to serum ACTH ratios >2 in basal condition or 3 after CRH stimulation are consistent with Cushing disease. Despite the very high sensitivity and specificity for this test, its use is limited by the cost and expertise necessary to perform the inferior petrosal sinus collection. Patients with ACTH-dependent Cushing syndrome without evidence of a pituitary cause are likely to have ectopic ACTH production from neuroendocrine malignancies. Therefore, in patients without the established cancer diagnosis, the next step would be to search for the occult malignancy with computed tomography scans of the chest and abdomen. The use of further imaging tests will be guided by the findings from the initial workup.
Treatment
Complete remission of Cushing syndrome may be achieved in the majority of patients where the ectopic source of ACTH is identified and the tumor is treated with curative intent. Nevertheless, curative resection is often not possible and patients require therapies aimed at decreasing the cortisol secretion by the adrenal glands. Although systemic therapy may improve the hypercortisolism, particularly in cases of SCLC, most patients require inhibitors of cortisol secretion. The most commonly used drugs are ketoconazole metyrapone and mitotane.114 Ketoconazole inhibits several steps in adrenal steroidogenesis, has a rapid onset of action, and is effective in approximately 50% of patients with ectopic ACTH secretion.115,116 The typical dose ranges from 400 to 1200 mg daily. Metyrapone inhibits 11-beta hydroxylase in the adrenal cortex, blocking the conversion of 11-deoxycortisol to cortisol. Mitotane is adrenolytic and inhibits several steroidogenic steps in the adrenal cortex. An alternative to medical therapy is bilateral adrenalectomy, which induces a rapid resolution of the Cushing symptoms and may be performed through laparoscopic surgery.117
Hypoglycemia
Hypoglycemia is most commonly caused by drugs, usually as a complication from treatment with insulin or oral hypoglycemic agents in patients with diabetes mellitus. Hypoglycemia in patients without diabetes mellitus undergoing treatment is rare and may be caused mainly by drugs, ethanol, liver disease, renal disease, congestive heart failure, endocrinopathies, malnutrition, sepsis, and malignancies.118
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


