Paraneoplastic Neurologic Syndromes
INTRODUCTION
Paraneoplastic neurologic syndromes are heterogenous disorders that can occur in the setting of various types of cancer (1). Paraneoplastic neurologic disorders are commonly caused by immune-mediated mechanisms triggered by an underlying tumor and have to be distinguished from neurological symptoms related to direct tumor invasion, infection, vasculopathy, ischemia, metabolic disturbances, or treatment-related toxicities.
Abnormal antibody or T-cell-mediated responses can target any part of the central, peripheral or autonomic nervous system to cause a diverse range of neurological symptoms. In general, the incidence of paraneoplastic syndromes is less than 1% in the general cancer population, but may be more frequently seen in specific types of cancer, such as small cell lung cancer (SCLC), cancers of the ovary and breast, and thymoma.
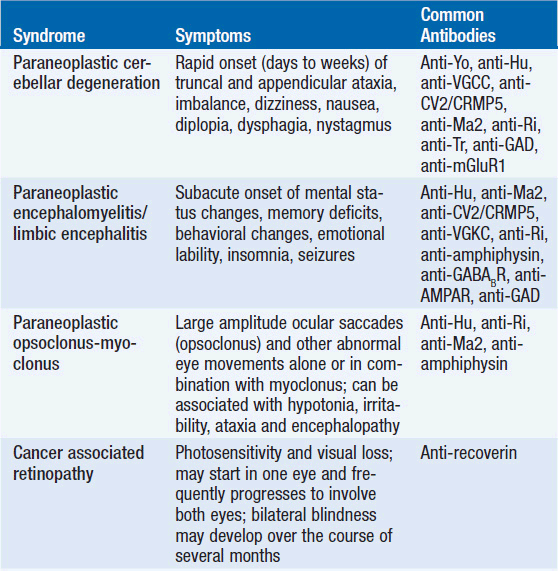
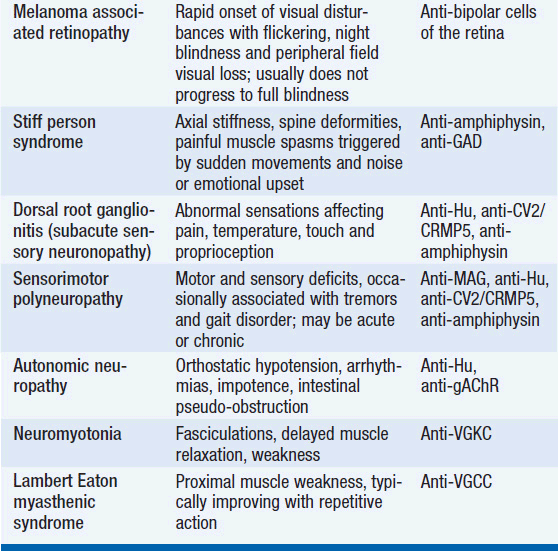
The diagnosis of a paraneoplastic neurologic syndrome is primarily clinical. Classical paraneoplastic syndromes (Table 64-1) may develop before the diagnosis of cancer, in a patient with known cancer, or in a patient considered to be in cancer remission. While an extensive search for an underlying tumor is warranted in classical paraneoplastic syndromes, such as paraneoplastic cerebellar degeneration (PCD) or Lambert-Eaton myasthenic syndrome, it may be noteworthy that several neurologic syndromes designated as “paraneoplastic” may also be seen in non-neoplastic autoimmune diseases.
Since paraneoplastic syndromes often herald the diagnosis of cancer, patients may be diagnosed and treated early in the course of the disease. Earlier cancer diagnosis may correlate with a higher likelihood of cure and remission of neurologic symptoms. It remains controversial, however, whether a better prognosis in patients with paraneoplastic syndromes is related to the immune control of the underlying cancer.
PATHOGENESIS
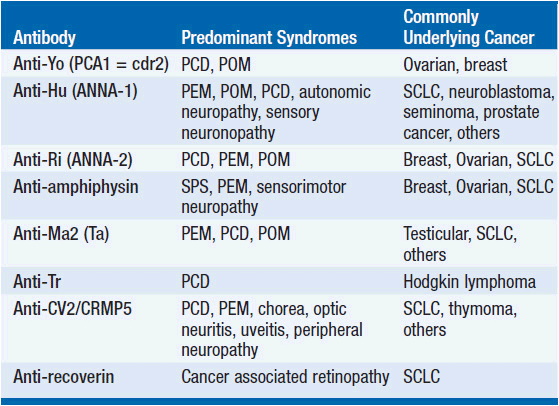
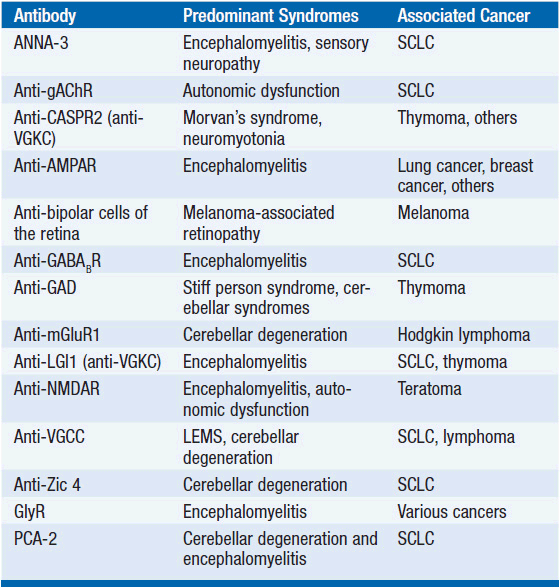
While the detailed pathomechanism of most paraneoplastic syndromes remains elusive, production of antibodies directed against antigens expressed by both tumors and nervous system tissues have been described as a key mechanism in some distinct types of paraneoplastic syndromes (Table 64-2). Antibodies can be directed against cytoplasmic antigens (e.g., Purkinje cells and anterior horn cells) or cell surface proteins (e.g., voltage gated potassium and calcium channels). Immune mechanisms may involve both humoral and T-cell-mediated responses, although the exact contribution of cellular and antibody-mediated mechanisms to the disease manifestation remains controversial (2–4). Abnormal antibodies can frequently be detected in serum and cerebrospinal fluid (CSF); however, a direct pathogenic effect of certain antibodies on nervous system tissue or the neuromuscular junction has only been demonstrated in a subset of paraneoplastic disorders. Importantly, some antibodies can also be detected in the absence of a clinical paraneoplastic syndrome (5) (Table 64-3).
TABLE 64-2 WELL-CHARACTERIZED PARANEOPLASTIC ANTIBODIES DIRECTED TO NON-SURFACE ANTIGENS, ASSOCIATION WITH NEUROLOGIC SYNDROMES AND UNDERLYING TUMORS
DIAGNOSTIC AND THERAPEUTIC CONSIDERATIONS
The diagnosis of a paraneoplastic neurologic syndrome can be challenging and, in classical syndromes, is usually based on clinical features of the neurologic condition, such as in cerebellar degeneration and Lambert-Easton myasthenic syndrome (1, 6, 7). Neurological symptoms commonly develop in the early stages of cancer and in two-thirds of patients before a cancer diagnosis has been established (1). Therefore, early recognition and treatment of a paraneoplastic neurologic disorder along with treatment of the underlying cancer are critical to reduce neurologic morbidity and to improve overall patient survival. Paraneoplastic neurologic syndromes usually have an acute or subacute onset, but the diagnosis may remain challenging if no typical antibodies are found in serum or CSF (7). As most syndromes and malignancies are associated with more than one specific antibody, screening for a panel of paraneoplastic antibodies may increase the yield to establish a diagnosis (7, 8). In patients with CNS manifestation, antibody screening in CSF is usually recommended, as antibody titers in CSF are generally higher than in serum (9). CSF analysis typically reveals an inflammatory pattern with mild pleocytosis, elevated protein, and evidence of oligoclonal bands. Positron emission tomography (PET) in combination with computed tomography (CT) is helpful in the search for an underlying malignancy (10, 11). Mammography and pelvic ultrasound may be indicated in women with suspected paraneoplastic neurologic syndromes, given the frequent association with breast and gynecologic cancers. Neuroimaging of the brain with magnetic resonance imaging (MRI) may be unremarkable but is abnormal in the majority of patients with encephalo-myelitis and limbic encephalitis (1). In cases in which no underlying tumor can be identified but a high clinical suspicion for a classic paranepolastic syndrome exists, or in patients tested positive for paraneoplastic antibodies in the setting of a less classic form of a paraneoplastic neurologic syndrome, repeat cancer screening studies are recommended every 6 months (7). An underlying malignancy usually can be identified within the first 4 years from onset of neurological symptoms (7).
The most important management goals in patients with paraneoplastic neurologic syndromes are identification and treatment of an underlying cancer. In general, antibody-mediated neurologic disorders affecting the peripheral nervous system will have a higher likelihood of response to treatment and symptom improvement. In these patients, immunomodulatory strategies with intravenous immunoglobulins, plasmapheresis, or immunosuppressants are commonly effective. While most paraneoplastic syndromes affecting the central nervous system generally respond poorly to immunomodulatory therapies, patients with opsoclonus-myoclonus syndrome (12) or limbic encephalitis and presence of anti-NMDAR, anti-AMPAR, or anti-Ma antibodies (13, 14) may show a remarkable benefit from treatment. In patients not actively treated with chemotherapy for an underlying cancer and with progressive neurological decline, more aggressive immunosuppression may be considered, including the use of cyclo-phosphamide, tacrolimus, azathioprine, and cyclosporine.
SYNDROMES OF THE CENTRAL NERVOUS SYSTEM
 PARANEOPLASTIC CEREBELLAR DEGENERATION
PARANEOPLASTIC CEREBELLAR DEGENERATION
Paraneoplastic cerebellar degeneration (PCD) is a classical paraneoplastic neurologic syndrome. Symptoms commonly precede tumor diagnosis by months or occasionally years and are characterized by rapidly progressive cerebellar dysfunction. The development of gait dysfunction, appendicular ataxia, and speech dysfunction may develop and progress over the course of days to weeks, unlike in primary degenerative cerebellar disorders, where symptoms develop over months to years. Brain stem and cerebellar dysfunction, including oculomotor symptoms with diplopia, oscillopsia, nystagmus, nausea, and dysphagia are usually irreversible (15, 16). Emotional lability and cognitive deficits may occur, underlining the importance of the cerebellum in mood and cognitive function (17). In addition, patients may develop pyramidal tract signs, posterior column signs, and polyneuropathy, suggesting a more widespread involvement of the nervous system in the pathology of the disease process. The majority of patients develop severe gait difficulties, inability to write and to swallow (18).
PCD is frequently associated with anti-Yo antibodies (Purkinje-cell antibody-1; PCA-1; also termed cerebellar degeneration related-2; cdr2), directed against Purkinje cells of the cerebellum, and usually occurs in patients with breast or ovarian cancer (16, 19). Because presence of anti-Yo antibodies is so frequently associated with underlying breast or ovarian cancer, careful and repeated cancer screening is mandated. Notably, more than two-thirds of patients with anti-Yo-associated cerebellar degeneration do not have a cancer diagnosis at the onset of neurologic symptoms. PCD has also been described in the presence of anti-Hu antibodies and anti-CV2/CRMP5 antibodies and SCLC (20), anti-Tr antibodies and Hodgkin lymphoma (21, 22), or anti-Ri antibodies and breast cancer (23). Interestingly, PCD has also been reported in patients with non-Hodgkin lymphoma and lung cancer without detectable anti-neuronal antibodies (24), and in SCLC patients with evidence of P/Q calcium channel antibodies (25).
As seen in other paraneoplastic neurologic syndromes, CSF analysis may reveal lymphocytic pleocytosis, elevated protein, and oligoclonal bands. At later disease stages neuroimaging with CT or MRI may identify loss of cerebellar architecture and eventually global cerebellar atrophy.
Treatment with plasmapheresis or intravenous immunoglobulins do usually not alter the progressive course of the disease (26), although clinical improvement has been seen in some cases (18, 27), with treatment of the underlying tumor (28, 29), or in some cases of PCD and underlying anti-Tr and anti-Ri antibodies (22, 30).
 PARANEOPLASTIC ENCEPHALOMYELITIS
PARANEOPLASTIC ENCEPHALOMYELITIS
Paraneoplastic encephalomyelitis (PEM) may affect multiple sites and levels of the nervous system, including the temporal and frontal lobes, midbrain, pons, cerebellum, spinal cord, dorsal root ganglia, and autonomic nervous system. Patients may therefore present with multiple neurological dysfunctions depending on the site of nervous system involvement. Symptoms usually develop over weeks to months. Involvement of the limbic system is characterized by short-term memory deficits, mood changes, emotional lability, and seizures. Brain stem involvement is classically associated with oculomotor dysfunction, nystagmus, and other cranial nerve palsies, whereas movement disorders are seen with midbrain involvement. Symptoms of autonomic dysfunction, such as cardiac arrhythmias, hypotension, and impaired gastrointestinal motility, can be identified in about 25% of patients. PEM typically occurs in the setting of lung cancer, particularly SCLC with presence of anti-Hu (ANNA-1) antibodies (20). Although less common, other antibodies have been linked to PEM, including anti-Ri antibodies (ANNA-2) in breast and gynecologic cancers (31), anti-CV2/CRMP5 antibodies in SCLC and thymoma (32), anti-amphiphysin antibodies in SCLC (33), anti-Ma2 antibodies in germ cell tumors and other cancers (34–36), and anti-NMDAR antibodies in ovarian teratomas (37, 38).
Diagnosis of PEM is usually clinical and supported by the presence of paraneoplastic antibodies. Neuroimaging studies with MRI frequently reveal T2/Flair signal hyperintensities of affected nervous system sites with and without gadolinium enhancement. In general, PEM is poorly responsive to treatment, although disease stabilization or mild symptom improvement has been reported with combined tumor treatment and immunotherapies, including intravenous immunoglobulins, plasmapheresis, or cyclophosphamide (38, 39).
 PARANEOPLASTIC LIMBIC ENCEPHALITIS
PARANEOPLASTIC LIMBIC ENCEPHALITIS
Paraneoplastic limbic encephalitis (PLE) is clinically associated with short-term memory deficits, mood disturbances, emotional lability, sleep alterations, behavioral abnormalities, and seizures (40). Symptoms may stabilize or improve over time; however, anterograde amnesia and cognitive deficits frequently persist. PLE typically occurs in the setting of SCLC with presence of anti-Hu antibodies, but can also be seen in presence of other antibodies, such as anti-Ma2, anti-NMDAR, anti-CV2/CRMP5, anti-amphiphysin, and anti-Ri (35, 38, 40–42). Notably, the clinical and radiographic picture of limbic encephalitis can also be seen in non-neoplastic autoimmune disorders characterized by presence of anti-VGKC (43, 44) and anti-AMPAR antibodies (14).
The diagnosis of PLE commonly precedes tumor diagnosis. Although less frequently, tumors other than SCLC have been associated with PLE, such as Hodgkin lymphoma, ovarian cancer, teratomas, testicular cancer, breast cancer, and thymomas.
Pathological findings may reveal perivascular and interstitial lymphocytic infiltrates and microglia activation in limbic structures, including hippocampus, amygdala, cingulate gyrus, and hypothalamus. There may be some overlap with the clinical picture of PEM, and neuropathology may not be restricted to limbic structures.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


