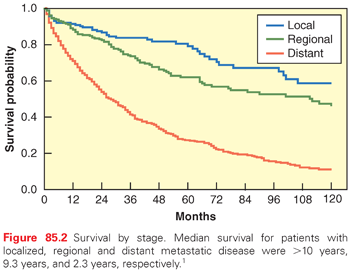
pNETs were slightly more common among men (53%) than women (47%), and the median age at diagnosis was 60 years.1,5 At diagnosis, 14% had localized disease, 22% had regional disease, and 64% had distant disease. The survival of patients with pNETs has improved over time.5,6 Among patients diagnosed from 1988 through 2004, the 5-year survival rates among patients with localized, regional, and distant pNETs were 79%, 62%, and 27%, respectively (Fig. 85.2).1
CLASSIFICATION, HISTOPATHOLOGY, MOLECULAR GENETICS
Criteria for Pathologic Diagnosis
A larger fraction of endocrine cells in the pancreas are insulin-producing (B) and glucagon-producing (A) cells. Relatively minor populations of cells produce somatostatin (D) and pancreatic polypeptide (PP). Rare cells also produce serotonin (EC) and ghrelin (P/D1).7 These endocrine cells are believed to be the source of pNETs. It is a matter of debate if tumors arise from islets or ducts. Transgenic mice expressing potent oncogenes in endocrine cells8 and multiple endocrine neoplasia (MEN)1 knockout mice9,10 point to an islet origin of tumors consistent with the auto-renewal properties of islet cells 11 and clinical observations in patients with MEN1.12 Conversely, molecular evidence from islet microdissection in patients with MEN1 indicate a duct cell origin.13 No matter where the truth lies, endocrine tumor cells largely display the same phenotype as their normal endocrine counterpart.
Aggressiveness of pNETs can be classified according to differentiation or proliferative rate. In the 2010 World Health Organization (WHO) classification, well-differentiated pancreatic neuroendocrine neoplasm is termed neuroendocrine tumor, whereas poorly differentiated neuroendocrine neoplasm is termed neuroendocrine carcinoma.14 Histologically, well-differentiated tumors are characterized by bland features: trabecular, glandular, acinar, or mixed structures; the stroma is generally fine and rich in well-developed blood vessels, sometimes with hyalinised deposits of amyloid; tumor cells are monomorphic with abundant, variably eosinophilic cytoplasm, low cytological atypia and low mitotic index. Necrosis is usually absent or may be seen as spotty, limited areas in histologically more aggressive neoplasms. On the contrary, poorly differentiated neuroendocrine carcinomas are characterized by prevalent solid structure with abundant necrosis, often central, round tumor cell of small to medium size with severe cellular atypia and high mitotic index.
Fine Needle Aspiration Versus Core Needle Biopsy
The diagnosis of pNET is necessary (1) to meet the previously defined histologic and cytologic criteria, (2) to assess the status of endocrine differentiation, and (3) to evaluate prognostic markers (proliferative index). Fine needle aspiration (FNA) biopsy is an effective technique in expert hands, allowing confirmation of the cytologic diagnosis on isolated or grouped cells with little information on tumor structure. The advantages of FNA are several, including its simplicity, low invasiveness, and cost. The disadvantages are mainly its operator-dependent efficacy and limited option for further study (small sample size) to include prognostic variables.
Conversely, the core needle biopsy (ideally 2 mm in diameter) produces a larger sized tumor sample, potentially allowing a cyto-/histologic diagnosis complete with all known prognostic parameters. Besides the easier diagnostic approach intrinsic to histology, its major advantage is certainly the potential for further studies to include all IHC studies and assessment of proliferative index. Disadvantages are the invasiveness of the procedure(s) and the relatively higher costs.
We typically recommend core needle when considering biopsy of liver metastases and FNA for biopsy of the pancreas.
Minimum Immunohistochemistry Markers
A large number of antigens, commonly defined as “neuroendocrine markers,” are expressed in tumor cells.7 They comprise markers dispersed in the cytosol including neuron-specific enolase (NSE) and protein gene product 9.5, and markers of the secretory compartment, either associated with electron-dense granules, large-dense-core vesicles such as chromogranins and related fragments (the most popular being chromogranin A [CgA]), or associated with small synaptic-like vesicles such as synaptophysin. These antigens are defined as “general markers,” since they are widely expressed in cells of the diffuse endocrine system. Hormones and/or amines are produced by specific cell types and thus defined as “specific markers.” The positive identification of the endocrine cell product(s) in tissue sections is obtained by IHC.
A minimal IHC histology panel is designed to (1) positively identify the degree of endocrine differentiation in tumor cells, and (2) determining the proliferation activity status. Determining the proliferation status is achieved by Ki67 IHC using the MIB1 antibody or it can be expressed as mitoses per 10 high power microscopic fields (or 2 mm2).
Grade—World Health Organization 2010, American Joint Committee on Cancer, Union for International Cancer Control
Proliferative rate has been widely acknowledged as an important prognostic factor in pNETs. Though some controversies remain, the grading system based on mitotic rate and Ki 67 labeling has been widely adopted by the American Joint Committee on Cancer (AJCC), International Union Against Cancer (UICC), and WHO (Table 85.1).15,16 Low (G1) and intermediate (G2) grade neoplasms are termed pNETs, whereas aggressive high-grade (G3) neoplasm are termed pancreatic neuroendocrine carcinoma.
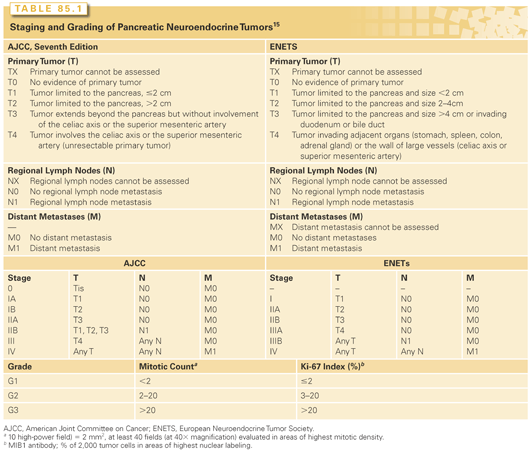
The general utility of such a grading scheme has been validated in a number of studies.17–21 However, the optimal Ki-67 cut-off between G1 and G2 remains debated in the literature. Some authors have proposed 5% (instead of 2%) as a more discriminatory cut-point for important outcomes including overall survival (OS).22 In one multivariate analysis that included lymph node ratio, a Ki-67 >5% was identified as the strongest predictor of recurrence after resection of malignant pNETs.23 At the current time, G1 and G2 tumors are managed in a similar way. Therefore, the distinction between G1 and G2, while prognostic, does not have major therapeutic implications. A more important issue is the distinction between G2 and G3 as platinum-based chemotherapy is generally recommended for G3 tumors. In a recent review of a large Nordic series, response rate to platinum-based chemotherapy was low for the subgroup of G3 patients with Ki-67 between 20% and 55%. Tumors with a Ki-67 >55% appear more responsive to platinum-based chemotherapy.24
Tumor, Node, Metastases (TNM)
The recent European Neuroendocrine Tumor Society proposal of tumor node, metastases (TNM) for non functional pNETs is based on criteria previously identified by the WHO 2004 classification and implies a malignant potential for any tumor type.15 The practical utility of this system was demonstrated in six independent studies.17–21,25 In 2010, a TNM system was officially provided by AJCC and the UICC.16 Their recommendation was to apply the same TNM system devised for exocrine pancreatic cancer to pNETs (see Table 85.1). The AJCC-UICC TNM scheme is based on a publication investigating a large tumor registry database.26 As there is currently no adjuvant therapy available for pNETs, the distinction between the two staging system carries no practical therapeutic implications.
Molecular Genetics of Pancreatic Neuroendocrine Tumors
Advances in technology over the past 5 years have led to an explosion of new data emerging from high throughput molecular analyses. Recent exome sequencing studies have simultaneously confirmed the importance of genes associated with inherited cancer syndromes and discovered important novel genetic aberrations among patients with sporadic pNETs.27
The importance of the MEN1 gene in the carcinogenesis of pNETs has long been implicated by the genetic cancer syndrome bearing its name as well as studies showing frequent loss of 11q13 where the MEN1 gene locus is found.28–33 Studies using comparative genomic hybridization and high-resolution allelotyping have demonstrated frequent amplification and deletions across a large number of chromosomes compatible with karyotypic instability.29–33 Exome sequencing studies identified three main groups (pathways) of mutations in sporadic pNETs. These include MEN1, DAXX or ATRX, and mammalian target of rapamycin (mTOR).27 MEN1 is thought to be involved in epigenetic regulation and is implicated in the regulation of endocrine mass during pregnancy through a p27-dependent pathway.34 DAXX and ATRX mutations are associated with alternative lengthening of telomeres and may offer escape from senescence.35 mTOR pathway mutations suggest an attractive therapeutic target that has now been validated in a pivotal phase III study.36
DIAGNOSIS AND MANAGEMENT OF PANCREATIC NEUROENDOCRINE TUMORS
Nonfunctional Tumors
Symptoms and Diagnosis
Nonfunctioning pNETs and PP-secreting tumors do not cause a clinical syndrome. Rarely, case reports of PP-secreting tumors have been associated with watery diarrhea,37–39 diabetes mellitus, ulcer diathesis,40 or an erythematous pruritic skin rash different from that seen in patients with glucagonomas.41 Until the tumor causes obstruction of the biliary tree or gastric outlet, the patients are usually asymptomatic unless the tumor bulk results in pain; this is in sharp contrast to pancreatic adenocarcinoma where very low tumor burden may be associated with profound symptoms and death. Jaundice may be the presenting symptom in patients with tumors to the right of the superior mesenteric artery (SMA) and superior mesenteric vein; these tumors originate in the pancreatic head or uncinate process, which may cause obstruction of the intrapancreatic portion of the common bile duct.42,43 Tumors in this location may also cause gastric outlet obstruction and/or pain owing to invasion of the autonomic mesenteric plexus. Pain may also be secondary to tumor extension into the celiac ganglion (most commonly seen with tumors arising in the body of the pancreas) or to liver metastases that invade the liver capsule or extend to the parietal peritoneum. Occasional patients may experience gastrointestinal hemorrhage secondary to tumor erosion into the duodenum or secondary to splenic vein occlusion causing gastroesophageal varices (sinistral portal hypertension). Nonfunctioning pNETs can sometimes grow to an enormous size without producing jaundice or other symptoms. pNETs arising to the left of the SMA and superior mesenteric vein may cause vague, poorly localized upper abdominal pain or dyspepsia, but such tumors are usually asymptomatic until they reach a considerable size. In contrast to patients with adenocarcinoma of the pancreas, patients with pNETs may not experience significant weight loss, cachexia, or back pain, or show other signs of advanced disease.42,43 In the absence of a large tumor or metastatic disease (of significant volume), pNETs are often detected incidentally on abdominal imaging studies.
On contrast-enhanced multidetector computed tomography (CT), pNETs characteristically appear hyperdense, as they are hypervascular. Therefore, imaging of the pancreas during the arterial phase is critically important to detect these lesions and their hypervascular liver metastases. However, similar to pancreatic adenocarcinomas, pNETs may occasionally appear hypodense compared with adjacent pancreatic parenchyma, and they may contain cystic components or microcalcifications. Importantly, intrapancreatic accessory splenic tissue can present as an asymptomatic, hypervascular mass involving the distal pancreatic tail, thus mimicking a pNET.
The current practice at our institutions is to obtain high-quality multidetector (multislice) CT images of the pancreatic tumor. We use objective CT criteria to determine a tumor’s resectability based upon the relationship of the pancreatic tumor to the SMA and celiac axis.44,45 Encasement (defined as >180-degree involvement of the vessel by tumor) of the SMA or occlusion of the superior mesenteric-portal venous confluence without the technical option of venous reconstruction are considered criteria for a tumor’s unresectability. Similar to our philosophy on local-regional management of pancreatic adenocarcinomas,46 we do not perform incomplete resection (debulking) of nonfunctioning pNETs. Some investigators have suggested that incomplete resection of the pancreatic tumor may provide relief of local tumor–related symptoms and improve survival, but most of these reports included patients with syndromes of hormone excess; no accurately reported data are available to support debulking in patients with unresectable nonfunctioning pNETs. However, in patients without distant metastases (or minimal liver metastases) extended resections to include complex vascular resection and reconstruction may be considered at those centers with experience in such complex operations.47,48 Magnetic resonance imaging (MRI) is preferred over CT for patients with a history of allergy to iodine contrast material or for those with renal insufficiency. Moreover, MRI may be more sensitive than CT for the detection of small liver metastases. Endoscopic ultrasound (EUS) is currently considered the most sensitive modality for identifying small pNETs and is thus used for preoperative tumor localization in patients with MEN1, in which multifocal disease is common. In the absence of an inherited endocrine syndrome associated with multifocal disease, the role of EUS is limited to FNA biopsy of the tumor. In the current era of invasive gastroenterology, EUS is safe and is becoming more widely available.
Accurate preoperative diagnosis and staging of the primary tumor is necessary to ensure correct treatment. The oncologic (surgical and medical) approach to a pNET is different from that for pancreatic adenocarcinoma. Because of the poor prognosis associated with pancreatic adenocarcinoma, patients with pNETs who are incorrectly thought to have large, locally invasive or metastatic adenocarcinomas may not undergo surgery when it is indicated and also may receive incorrect chemotherapy. Unless a surgery first strategy is considered (in which preoperative biopsy may not be necessary) pretreatment FNA biopsy should always be performed to prevent an error in diagnosis; this is especially true when the clinical history, physical examination, and radiographic images are confusing and not consistent with the presumed clinical diagnosis.
Serum Tumor Markers
Several circulating tumor markers have been evaluated for the diagnosis and follow-up management of pNETs. While these can be very useful for follow-up, isolated elevation of marker levels is generally not sufficient for diagnosis. These markers usually can be divided into those associated with specific endocrine syndromes and those more general markers that may be present in functional as well as nonfunctional tumors. The most important of these markers, CgA, is a 49-kDa acidic polypeptide that is widely present in the secretory granules of neuroendocrine cells. CgA is elevated in the majority of patients with either functioning or nonfunctioning pNETs.49–52 In a study where patients with advanced pNETs were treated with streptozocin-based chemotherapy, 79% of patients had elevation of CgA at the time of diagnosis.53 In addition, response to therapy was associated with a 30% decrease in serum CgA.53 This concept was also tested in the RADIANT-1 study, which treated patients with progressive pNETs after cytotoxic chemotherapy with the mTOR inhibitor everolimus. In this study, a 30% decrease in CgA, 4 weeks after initiation of therapy, was associated with significantly longer progression-free survival (PFS).54 However, care should be taken in measuring CgA and interpreting the results. For example, as somatostatin analogues are known to affect blood levels of CgA, serial CgA levels should be measured at approximately the same interval from injection in patients receiving long-acting somatostatin analogues. Spuriously elevated levels of CgA have also been reported in patients using proton pump inhibitors, in patients with renal or liver failure, or in those with chronic gastritis.
Another general neuroendocrine marker, NSE, is a dimmer of the glycolytic enzyme enolase. NSE is present in the cytoplasmic compartment of the cell, and its serum level is thought to be unrelated to the secretory activity of the tumor.52 While less specific as a diagnostic marker, it may be helpful in the follow-up of patients with unresectable disease. In the RADIANT-1 study, a 30% decrease in NSE at week 4 was associated with significantly longer PFS.54
PP levels also are frequently elevated in patients with pNETs. Elevation of PP is not associated with a distinct hormonal syndrome and is only considered significant when PP level is at least three times the age-matched normal basal level obtained in a fasting state.55 A variety of other secreted amines can also be measured. These include other chromogranins such as chromogranin B and C, pancreastatin, substance P, neurotensin, neurokinin A, gastrin, glucagon, vasoactive intestinal peptide, insulin, proinsulin, and c-peptide. In general, blood markers should be drawn in the fasting state.
It is recognized that NETs sometimes can change what (if any) hormones and biomarkers are produced. The general principle of biomarker measurement is to evaluate a large panel of markers at key points in time (diagnosis or relapse) in order to identify the biomarkers that are elevated and then follow these over time. It is generally not necessary to check every biomarker at every visit.
Surgical Treatment
The majority of pNETs are malignant, with the exception of insulinomas, which are benign in 95% of patients. It is probably best to assume that if left untreated, all non–insulin-secreting pNETs have the biologic ability for uncontrolled local growth and metastasis to distant organs. pNETs frequently metastasize to regional lymph nodes, and the frequency of lymph node metastases depends on the extent of surgery and on the degree and accuracy of the pathologist’s examination of the surgical specimen.56
Based on our experience and the reports from others, we have developed general guidelines for the surgical management of patients with nonfunctioning pNETs:
1. We establish the diagnosis with needle biopsy (EUS-guided FNA of the pancreas or image-guided core needle biopsy of the liver metastases is preferred) and decompress the biliary tree with an endobiliary stent if a distal bile duct obstruction is present. Once biliary obstruction is recognized and a stent is placed in the bile duct, an operation to remove the primary tumor or bypass the site of obstruction will likely be needed; in the absence of large volume distant metastases, the patient will most likely outlive the biliary stent and experience significant stent-related morbidity.
2. We resect localized, nonmetastatic disease confined to the pancreas if a gross complete resection can be performed. If radiographically occult liver metastases are found at the time of the operation, they are removed if possible. If the liver metastases are of small volume but diffuse, the primary tumor is usually removed due to the potential for major morbidity from the primary, which is a possibility because of the relatively long-anticipated survival of the patient.
3. There is an emerging body of literature to suggest that nonfunctioning pNETs < (approximately) 2 cm in diameter are of limited metastatic potential. This is especially important when dealing with patients of advanced age or clinically significant comorbidities (which increase operative risk even if the procedure is performed laparoscopically). In this setting, if the pNET represents an incidental finding on CT/MRI obtained for an unrelated reason, observation may be the best approach.57–59 Repeat imaging in 6 to 12 months will provide a window of opportunity to assess tumor biology. If observation is chosen and the diagnosis is confirmed on imaging, to include a functional study such as somatostatin receptor scintingraphy, biopsy may not be necessary.
4. In the setting of known metastatic disease or a large, borderline resectable primary tumor, we would first initiate systemic therapy as a bridge to eventual operation. Significant downstaging of the overall tumor burden can improve the safety of surgery in some patients.
5. The decision to operate on the primary pancreatic tumor is based upon the presence and/or extent of distant disease and the presence or absence of symptoms (bleeding, obstruction) from the primary tumor. For example, resection of an asymptomatic primary in the distal pancreas has a limited role, if any, in the presence of unresectable, moderate- to large-volume extrapancreatic metastatic disease. As treatments for metastatic disease become more effective, the rationale for aggressive management of the primary tumor despite the presence of extrapancreatic disease may become more compelling. However, treatment sequencing will likely emphasize a surgery-last strategy (after induction systemic therapy) to identify those patients most likely to benefit from large, multiorgan resections.
6. When dealing with a resectable primary tumor and resectable liver metastases, we usually remove the pancreatic tumor first; if that procedure goes well, we then consider resecting the liver under the same anesthesia induction.60 However, we often use a two-stage procedure if all needed surgery cannot safely be performed at one operation.
The goals of surgery are to maximize local disease control and to increase the quality and length of patient survival. These goals must be tempered by the potential operative morbidity and the long-term complications of insulin dependence and gastrointestinal dysfunction. We previously reported survival data for 163 patients with nonfunctioning pNETs treated at our institution.61 As expected, patients with localized or regional disease at diagnosis had a significantly superior median survival compared with those who had metastatic disease (7.1 years versus 2.2 years; p <0.0001). Among patients with localized disease, those who underwent complete resection of the primary tumor demonstrated an additional survival advantage over those with locally advanced, unresectable tumors (median survivals of 7.1 years for patients with localized, resectable disease versus 5.2 years for patients with locally advanced, unresectable tumors).61 However, only 48% of the 42 patients with localized, nonmetastatic disease who underwent resection of the primary tumor were alive and without evidence of recurrent disease at a median follow-up of 2.7 years (range, 1 to 8 years) from diagnosis. It is thus inappropriate to assume that complete resection of the primary tumor in the absence of metastatic disease corresponds to long-term cure.
Occasionally, an extended operation is required to achieve complete tumor resection of nonfunctioning pNETs. A high-risk operation (to include most that require complex vascular resection and reconstruction) should not be performed in a high-risk patient who because of age and medical comorbidities has a significant risk of perioperative mortality (=10%) or morbidity (=30%). In the absence of surgery, survival duration is often measured in years even in the presence of distant metastases and therefore surgery-related complications are to be avoided.62 At present, we are highly selective in our use of extended pancreaticoduodenectomy or left-sided resections to include the celiac axis (Appleby procedure). Patients who undergo such high-risk operations must have limited to no medical comorbidities and have an excellent performance status. Decisions for or against surgical treatment are particularly difficult when dealing with large primary tumors, which require an extend resection, in the absence of distant metastases. The median survival duration for patients with unresectable, nonmetastatic, nonfunctioning pNETs is approximately 5 years. As survival time without operation increases and as potential operative morbidity and mortality increase, we are less accepting of the upfront risks of surgery. However, as mentioned previously, occasionally, locally advanced tumors of the pancreatic head or uncinate process are associated with significant patient morbidity due to complications such as biliary obstruction, gastric outlet obstruction, or gastrointestinal hemorrhage.61 In contrast to the management of patients with pancreatic adenocarcinoma (where endoscopic stenting of the bile duct and occasionally the duodenum are fairly routine in the setting of locally advanced or metastatic disease), we would rarely use a duodenal stent in a patient with neuroendocrine carcinoma and would utilize endobiliary stents only for short-term (months, not years) biliary decompression. Because of the longer survival times of patients with pNETs (even advanced disease), we favor operative bypass of the bile duct and duodenum in most cases.
Oncologic Management of Advanced Pancreatic Neuroendocrine Tumors
Advanced, unresectable pNETs generally are not curable. The goals of oncologic management include palliation or prevention of symptoms and cytoreduction of bulky tumors in an effort to prolong survival. Although low- to intermediate-grade pNETs have a reputation of being indolent, most patients with advanced pNETs will not survive the disease.
Management of patients with advanced pNETs requires an understanding of the disease process and the importance of a multimodality approach. Treatment options include cytotoxic chemotherapy, everolimus, sunitinib, somatostatin analogues, peptide-receptor radiotherapy, as well as ablative approaches such as hepatic artery embolization and radiofrequency ablation (RFA). Occasionally, systemic therapy may also convert cases of unresectable tumors into cases wherein surgery may render the patients disease free. In such cases, we recommend that surgical options be considered in a multidisciplinary setting. Much of what is discussed here for non functional pNETs also holds true for managing the growth of functional tumors.
Systemic Therapy
Somatostatin Analogues
Somatostatin is a hormone that binds to specific high-affinity membrane receptors on target tissues. To date, five subtypes of somatostatin receptors (SSTR) have been identified. When activated, these receptors trigger differing biologic activity. The somatostatin analogues octreotide and lanreotide both bind with high affinity to SSTR 2 and with slightly lower affinity to SSTR 5. Pasireotide is a novel cyclohexapeptide in development that binds to SSRT 1, 2, 3, and 5.63
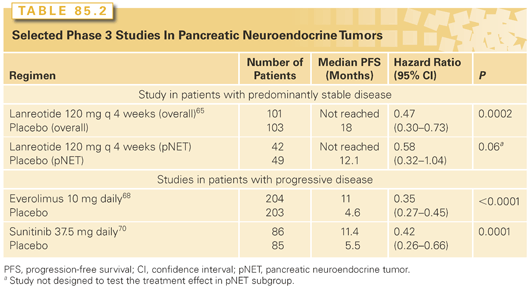
The somatostatin analogue octreotide is approved for the control of symptoms related to hormonal hypersecretion in NETs. More recently, randomized controlled studies have also demonstrated that somatostatin analogues can delay tumor growth. The PROMID study compared octreotide LAR 30 mg every 4 weeks to placebo among treatment-naïve patients with midgut NETs (carcinoid tumors). The study demonstrated significant benefit in time to progression (hazard ratio [HR] = 0.34; 95% CI, 0.20 to 0.59; p <0.0001).64 In a larger study that included a substantial number of pNETs, lanreotide 120 mg was compared to placebo in a population of patients with mostly stable disease (Table 85.2). Treatment with lanreotide was also associated with significant benefit in PFS (HR = 0.47; 95% CI, 0.30 to 0.73; p = 0.0002).65 The long median PFS of 12 (95% CI, 9.4 to 18.3) months among patients with pNET receiving placebo suggests that many patients with known stable disease may not need immediate treatment.
Everolimus
mTOR is an intracellular protein that has a central role in cellular function. It acts as a nutrient sensor and mediates signaling downstream of receptor tyrosine kinases controlling cell growth, protein synthesis, autophagy, and angiogenesis. The association between aberrant mTOR pathway signaling and the pathogenesis of pNETs is suggested by the development of pNETs in patients with inherited genetic mutations in TSC2 and NF1. Loss of TSC2 and NF1 are both associated with mTOR activation. In an exome sequencing study, mTOR pathway mutations were also found in sporadic pNETs.27 Finally on a protein level, low expression of PTEN and TSC2 were associated with poor prognosis.66
Phase 2 studies of the mTOR inhibitor everolimus have reported evidence of clinical activity.54,67 In the initial report, the combination of octreotide LAR and everolimus was studied in 60 patients with NETs.67 The response rate among 30 patients with pNET was 27%. In a subsequent multinational phase 2 study (RADIANT-1) in advanced pNETs with progression following chemotherapy, 160 patients were treated in two strata, with everolimus (n = 115) or everolimus plus octreotide (n = 45) based on whether patients were on octreotide at study entry.54 By central radiology review, the response rate was lower at 9.6%. Durable disease stabilizations were, however, observed among patients with progression at study entry. The median PFS for patients receiving everolimus or everolimus plus octreotide were 9.7 months and 16.7 months, respectively. In the largest phase 3 study to have been conducted in pNETs (RADIANT-3), 410 patients with progressive pNETs were randomly assigned to receive everolimus or placebo. The study demonstrated clinically and statistically significant benefit in PFS for patients receiving everolimus (see Table 85.2).68 Everolimus prolonged median PFS from 4.6 months to 11 months leading to a 65% risk reduction for progression compared to placebo (HR = 0.35; 95% CI, 0.27 to 0.45; p <0.0001). Treatment also reduced the level of tumor-secreted hormones.
Sunitinib
pNETs are vascular tumors known to express vascular endothelial growth factor (VEGF). Recent studies have demonstrated the expression of VEGFR-FLK and VEGFR-FLT1 on tumor cells. Sunitinib is a novel tyrosine kinase inhibitor with activity against VEGF receptors, c-Kit, and platelet-derived growth factor receptor. In a multicenter phase 2 study, investigators treated patients with carcinoids and pNETs in separate strata and also observed evidence of clinical activity. Interestedly, in this study, the tumor response rate appeared to be higher among patients with pNETs than in patients with carcinoids (17% versus 2%).69 A subsequent phase 3 study compared sunitinib to placebo in pNETs (see Table 85.2). Results of an early unplanned analysis showed improved PFS (5.5 months versus 11.4 months).70 Although the study showed clinically meaningful benefit (HR = 0.42, 95% CI, 0.26 to 0.66), the type 1 error was uncontrolled. Due to the small number of events and unplanned nature of the analyses, the results failed to cross the O’Brian Fleming efficacy threshold for statistical significance.
Cytotoxic Chemotherapy
Systemic chemotherapy for advanced pNETs has been studied in many clinical trials over the last three decades. Despite the multitude of publications, the role of cytotoxic chemotherapy continues to be debated. Older studies often used criteria to measure outcome that are not accepted today. Older studies using cytotoxic agents have not documented improvements in PFS or OS versus best supportive care.71,72
Streptozocin-Based Chemotherapy. Streptozocin was originally isolated from streptomyces achromogenes in the 1950s. Its antitumor activity in pNETs was first reported in 1973; in a study that included 52 patients, a response rate of 50% was reported.73 Streptozocin’s single-agent activity in pNETs was subsequently confirmed in a study comparing that agent alone with streptozocin plus fluorouracil.71 In this study, a higher response rate was reported for the combination of streptozocin and fluorouracil. The Eastern Cooperative Oncology Group subsequently compared this combination to streptozocin plus doxorubicin72 and reported a significantly higher response rate (69% versus 45%), time to progression (median, 20 months versus 7 months), and OS (median, 2.2 years versus 1.4 years) for streptozocin plus doxorubicin than for streptozocin plus 5-FU. Based on these data, combination chemotherapy with streptozocin-based regimens is considered the standard treatment option by many.
However, two small retrospective series have recently cast doubt on the value of streptozocin-based chemotherapy. Each of these studies examined only 16 patients. Both reported a disappointing radiologic response rate of only 6%.74,75 This 10-fold difference in response rates has aroused considerable controversy as to the role of chemotherapy in treating pNETs. Some of the disparity in response rate may be accounted for by differences in response criteria. For example, in a study reported by Eriksson et al.,76 the response rates based on either decreased biochemical parameters or decreased tumor measurement were 36% for streptozocin plus doxorubicin and 58% for streptozocin plus 5-FU. When only radiologic response was counted, the respective response rates were 8% and 32%.76
A chemotherapeutic combination of 5-FU, streptozocin, and doxorubicin was studied in two small trials with 10 and 12 patients, and response rates were 40% and 55%, respectively.53 In light of the continuing controversy regarding the role of chemotherapy in the management of pNETs, a larger retrospective study examined the outcome of 84 consecutive patients treated with the 5-FU-doxorubicin-streptozocin combination and observed a response rate of 39%.53 The median PFS in that series was 18 months, and median OS was 37 months.
Dacarbazine- and Temozolomide-Based Chemotherapy. Dacarbazine was initially studied in a phase 2 study that included 42 patients with pNETs. A response rate of 33% was observed.77 Temozolomide is an oral alkylating agent that metabolizes to the same active metabolite as dacarbazine, 5-(3-methyl-triazeno) imidazole-4-carboxamide. While a number of temozolomide-based doublets have been reported in clinical trials or retrospective series, the activity of single-agent temozolomide has not been prospectively evaluated.18–20 In one large series, 18 (34%) of 53 patients with pNETs had objective response following temozolomide-based chemotherapy.78 Although temozolomide-based therapy is generally well tolerated, absolute lymphopenia may occur and has been associated with opportunistic infections.79 These studies suggest that temozolomide may have activity in pNETs. A randomized study comparing temozolomide versus temozolomide plus capecitabine is ongoing.
Cytotoxic chemotherapy continues to play an important role in the management of pNETs.
Peptide Receptor Radiotherapy
The presence of somatostatin receptors in high density on tumors cells has led to the development of peptide receptor radiotherapy for NETs. Early studies with 111In-, 90Y-, or 177Lu-labeled somatostatin analogues have reported promising results in the control of hormone-associated symptoms.80
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


